





Malattia genetica della retina classificata tra le degenerazioni tappeto-retiniche, che provoca cecità totale o parziale fin dall’infanzia (in genere nei primi sei mesi di vita).
È la causa più frequente di cecità infantile ereditaria, con un’incidenza di 3 casi ogni 100.000 nati vivi.
I sintomi tipici sono la marcata ipovisione ed il nistagmo, cioè il movimento continuo e incontrollato degli occhi.
Non esiste una terapia definitiva ma negli ultimi anni la somministrazione del gene terapeutico tramite un vettore virale inoculato direttamente nell’occhio dei pazienti si è rivelata una strategia promettente per una forma di questa patologia.
L’amaurosi transitoria (o fugace), invece, è un episodio di cecità transitoria causata da ischemia retinica o del nervo ottico, causata dal restringimento dell’arteria carotide e riscontrabile più frequentemente nei pazienti ultra cinquantenni.
L’ostruzione dell’arteria carotide sinistra è sei volte più frequente di quella dell’arteria carotide destra. I blackout si presentano come offuscamento della visione in un occhio con un lento recupero che inizia dopo 5–10 minuti.
La visione chiara sarà ripristinata in ordine inverso dal quadro di insorgenza.
Diversi episodi di blackout possono precedere un attacco di neuropatia ottica ischemica oppure possono verificarsi episodi per anni senza gravi sequele.
I blackout possono anche essere bilaterali, se associati a pressione arteriosa bassa.
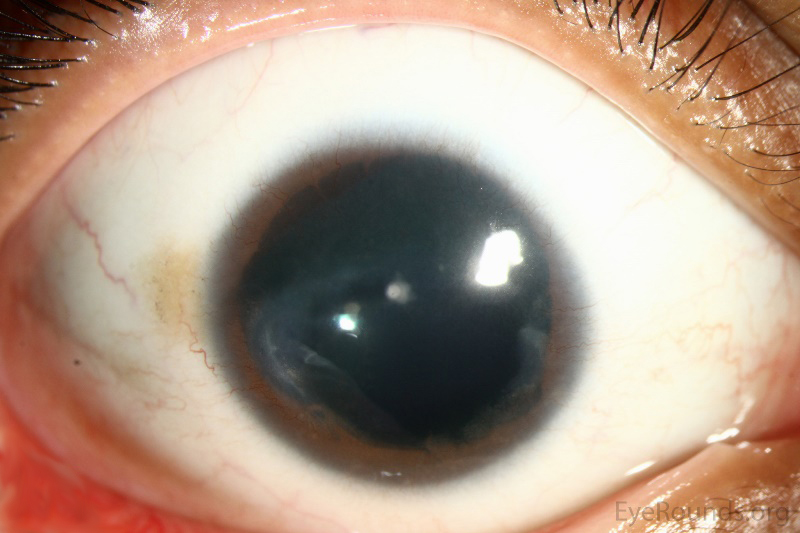
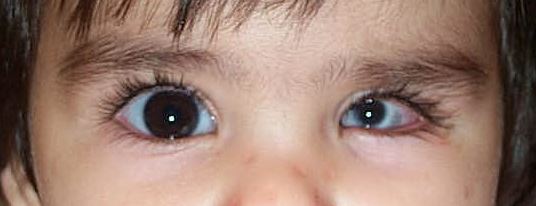
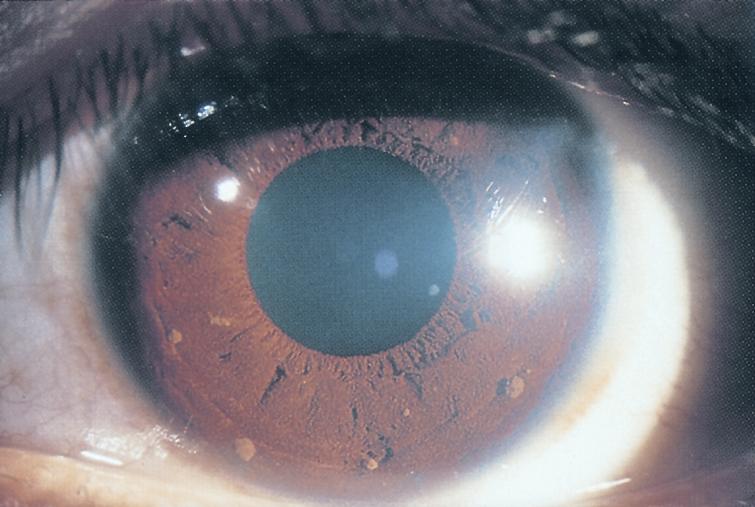
Ipoplasia o assenza completa dell’iride di origine congenita causata da una mutazione del gene PAX6, situato sul cromosoma 11, che determina un mancato completamento dello sviluppo dell’occhio. Colpisce da 1: 40.000 a 1: 100.000 nati vivi, maschi e femmine in ugual misura. Si presenta con la mancanza di tessuto pigmentato dietro la cornea e dà l’impressione di un’enorme dilatazione della pupilla.
La malattia è spesso trasmessa come carattere autosomico dominante da genitori portatori della mutazione (aniridia ereditaria). Un terzo dei casi può invece derivare da una mutazione genetica che si verifica in un bambino i cui genitori non sono portatori della malattia (aniridia sporadica). Alcuni pazienti affetti da aniridia possono essere colpiti anche da patologie associate come fotofobia, nistagmo, glaucoma, cataratta e cheratopatia.
L’aniridia può manifestarsi nel quadro della Sindrome di Wagr una malattia genetica che colpisce più di un gene sul cromosoma 11 e la mutazione può variare da paziente a paziente. La sindrome può provocare tumore di Wilms (tumore renale infantile), anomalie genito-urinarie, aniridia e ritardo mentale.
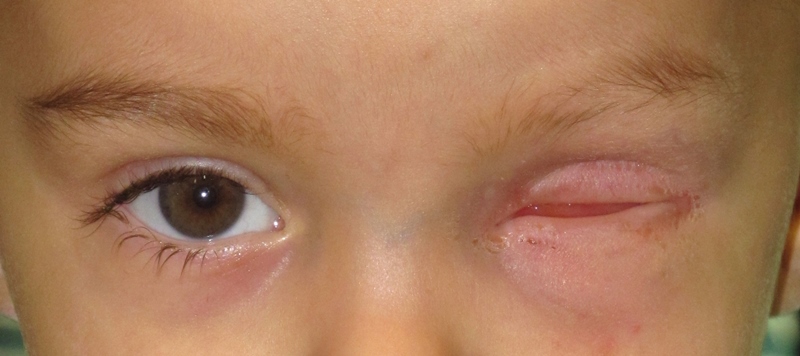
L’anoftalmia o anoftalmo è una condizione fisiologica che consiste nella totale assenza del bulbo oculare mentre sono presenti gli annessi oculari, cioè palpebre, congiuntiva e l’apparato lacrimale.
Questa condizione può essere congenita e in questo caso è dovuta alla mancata formazione della vescicola ottica tra la quinta e la sesta settimana di vita intrauterina: è una malformazione molto rara, con un’incidenza di 0,8 su 100.000 nati vivi, a volte bilaterale e senza preferenza di sesso e può essere isolata o associata ad altre malformazioni. Nell’anoftalmia congenita è presente un’ipoplasia emifacciale (se monolaterale) o nei casi bilaterali un’ipoplasia facciale completa.
I bambini nati con questa malformazione presentano in genere malformazioni palpebrali che possono risultare non perfettamente formate.
L’anoftalmia congenita può essere suddivisa in tre forme distinte:
Oltre che congenita, la condizione di anoftalmia può essere il risultato di un intervento chirurgico causato per eventi di natura traumatica o per altre forme patologiche. L’anoftalmia chirurgica è infatti la conseguenza dell’asportazione del bulbo oculare (enucleazione) o del suo contenuto, con conservazione del guscio sclerale (eviscerazione) a causa di gravi patologie acute come l’endoftalmite o il glaucoma oppure neoplastiche come il melanoma della coroide e retinoblastoma, o ancora per effetto di traumi orbitali importanti che hanno reso inevitabile la rimozione del bulbo oculare.
La diagnosi di anoftalmo viene seguita da un percorso di riabilitazione protesica con l’applicazione di una protesi oculare, di norma composta di resina acrilica anallergica (un materiale inerte e biocompatibile utilizzato nella chirurgia) che deve essere eseguita su misura del paziente per rispettare la sua morfologia e, nel caso di pazienti pediatrici, per favorire il corretto e simmetrico sviluppo dell’apparato osseo e tissutale della regione orbitale, in armonia con l’orbita controlaterale.
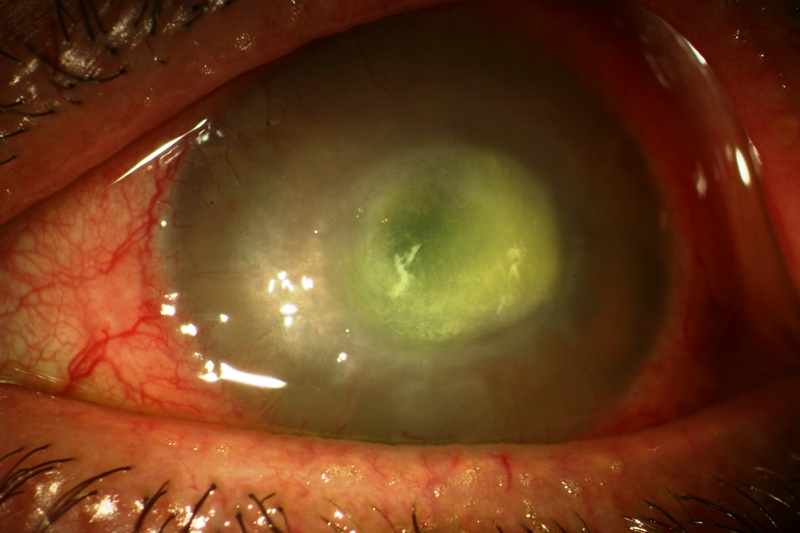
E’ una cheratite profonda con raccolta di pus localizzata all’interno degli strati più profondi della cornea, cioè un processo flogistico corneale che porta a morte i tessuti interessati. E’ spesso dovuto ai postumi di ferite o altre lesioni perforanti ed inizia con un’infiltrazione della parte centrale della cornea a cui fa seguito l’interessamento dell’uvea; questa zona assume una colorazione giallastra. Il materiale purulento sporge verso la camera anteriore in cui può aprirsi determinando un’ulcera posteriore della cornea. Se non si interviene con un energico trattamento a base di antibiotici e atropinici, si può perforare la cornea con la conseguente penetrazione dei batteri all’interno del bulbo che può provocare una panoftalmite, con il rischio della perdita del bulbo stesso.
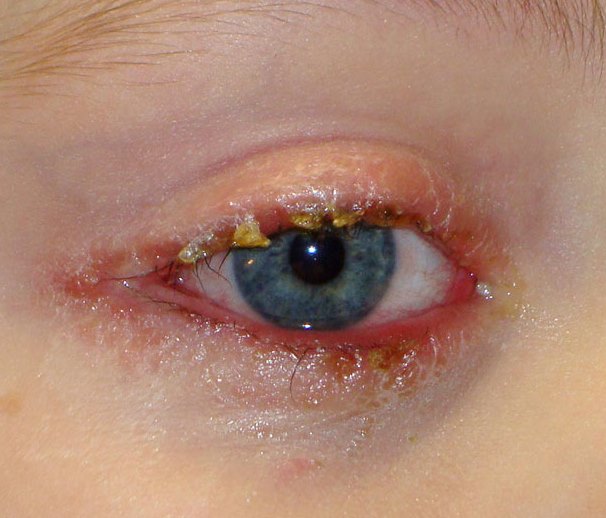
Infiammazione delle palpebre, in particolare dei bordi. Le palpebre diventano edematose e sui bordi compaiono piccole bolle e squamette. Le ciglia possono cadere ed è presente abbondante secrezione purulenta, tanto che le palpebre al risveglio rimangono incollate. E’ un disturbo molto diffuso e con caratteristiche recidivanti e contagiose.
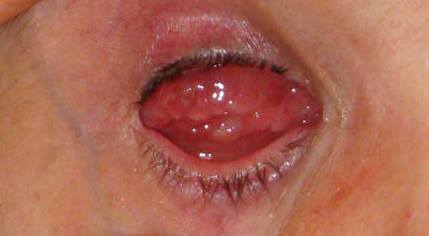
Sinonimo di sinechia, è costituita da cordoni o lamine fibrose di varia lunghezza e spessore e che possono essere causa di disturbi funzionali, la cui gravità è in rapporto con l’organo interessato e con l’estensione e il carattere della briglia stessa.
Le briglie sono costituite da tessuto connettivo e la loro formazione è preceduta sempre da essudazione fibrinosa che unisce le parti, poi si neoforma un connettivo giovane che sostituisce la fibrina: questa viene poi riassorbita ed il connettivo giovane si trasforma in connettivo fibroso che si compatta, presentando a volte infiltrazioni calcaree e qualche volta vere e proprie ossificazioni.
Le briglie cicatriziali di grosse dimensioni presenti nella cavità anoftalmica possono rendere particolarmente fastidiosa se non impossibile l’applicazione della protesi oculare.
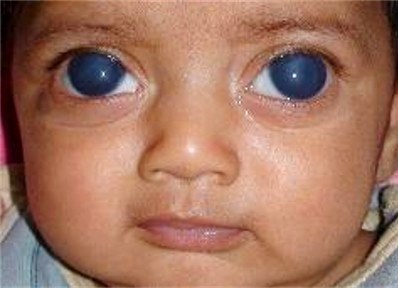
Condizione in cui il bulbo presenta dimensioni aumentate a causa dell’ elevata pressione intraoculare nei pazienti affetti da glaucoma congenito. Il bulbo esoftalmico è spesso associato a progressivo danneggiamento della papilla ottica e ad opacamento corneale.
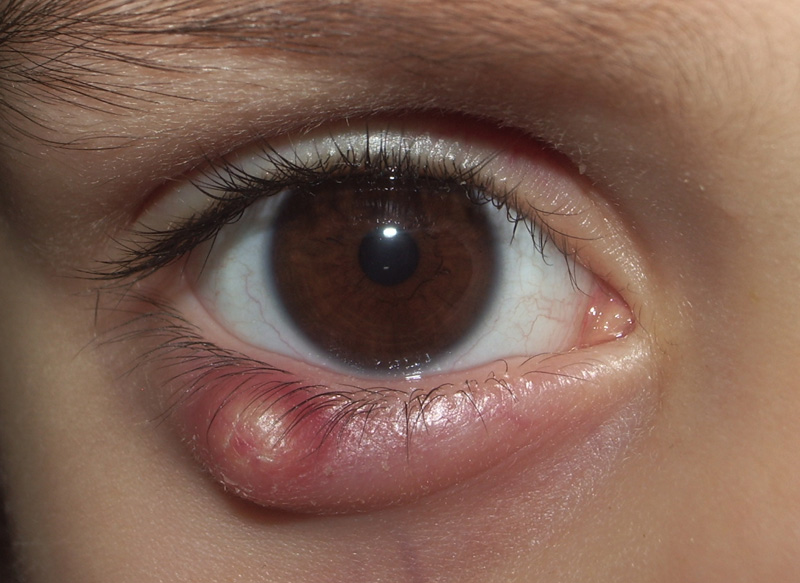
Cisti granulosa (meglio definita lipogranuloma) che si forma nella palpebra a causa di un’infiammazione cronica della ghiandola di Meibomio il cui dotto escretore si ostruisce. La dilatazione della ghiandola produce la proliferazione connettivale che si trasforma in un piccolo granuloma e può andare incontro a suppurazione batterica. In questo caso deve essere asportato chirurgicamente incidendo la capsula e rimuovendo il contenuto infetto.
In base alla sua posizione, vicino al margine ciliare o più verso la congiuntiva si definisce rispettivamente calazio esterno o interno, anche se in alcuni casi possono sussistere entrambi contemporaneamente (si parla quindi di calaziosi).
Il calazio si riconosce per il rigonfiamento all’interno o sul bordo della palpebra ed è generalmente indolore. In alcuni casi la ghiandola di Meibomio si svuota e il calazio guarisce da sé ma in situazioni più gravi può associarsi ad una blefarite che va contrastata da un’antibioticoterapia.
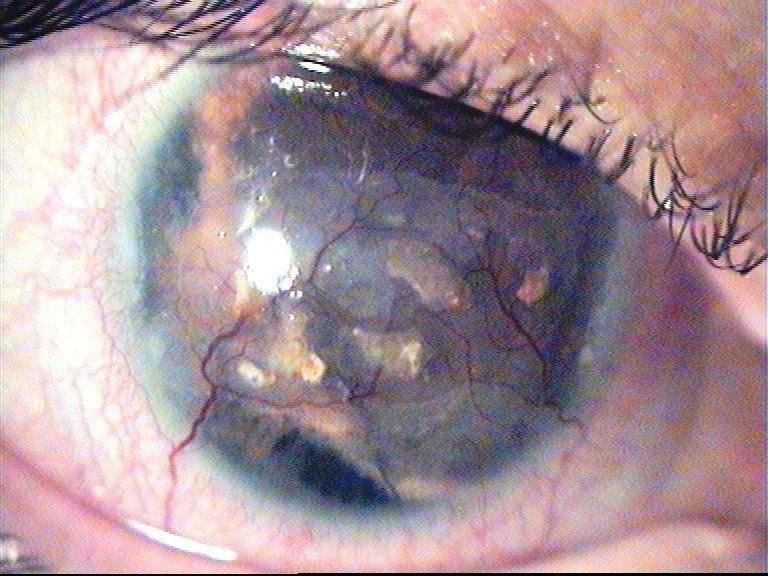
Tipica complicanza della sindrome dell’occhio secco, della cheratopatia a bandelletta calcifica e riscontrabile in genere nell’ipercalcemia che consiste nell’accumulo di calcio che può depositarsi nello stroma superficiale, nella membrana di Bowmann e negli strati profondi dell’epitelio.
Le calcificazioni sono frequentemente riscontrabili nella ftisi del bulbo.
Se la calcificazione si forma nella parte centrale della cornea può ovviamente interferire con la visione e può essere anche dolorosa.
I pazienti affetti da queste patologie, nelle forme più avanzate, vanno incontro a questa complicazione ed anche alla cheratopatia striata, un’alterazione dolorosa della superficie della cornea.
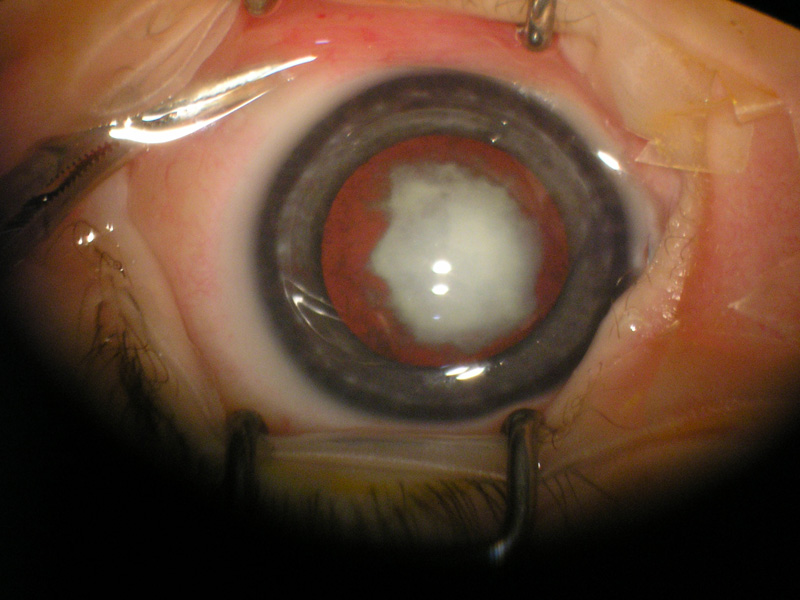
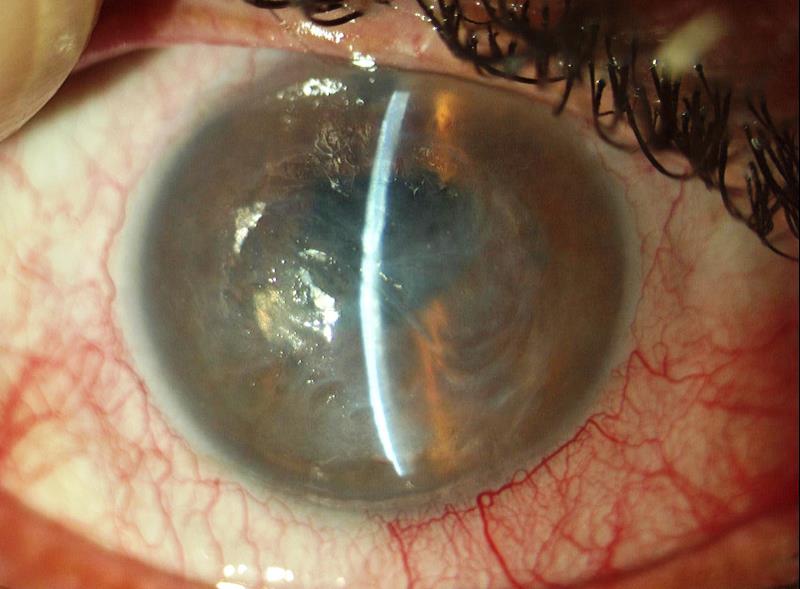
Opacizzazione del cristallino che può essere congenita o svilupparsi progressivamente in età senile. Il sintomo principale è l’abbassamento del visus e l’annebbiamento della vista. Il cristallino catarattoso viene di norma rimosso chirurgicamente e sostituito con un cristallino artificiale (IOL) che ripristina in toto la sua funzione. Le cause possono essere:
Quando l’opacità è localizzata nel nucleo centrale del cristallino (cataratta nucleare) nelle prime fasi si sviluppa la miopia che consente ad un paziente presbite di leggere senza correzione.
Se l’opacità è situata a livello della capsula posteriore del cristallino (cataratta sottocapsulare posteriore) influenza negativamente la vista ed è particolarmente fastidiosa se il paziente è esposto alla luce molto forte.
L’intervento di cataratta è raccomandato quando il visus massimo corretto è uguale o inferiore a 4/10 o quando la menomazione soggettiva visiva impedisce attività necessarie come la guida o la lettura. Anche l’abbagliamento disabilitante può costituire un’indicazione all’intervento ed è più comune nelle cataratte sottocapsulari posteriori.
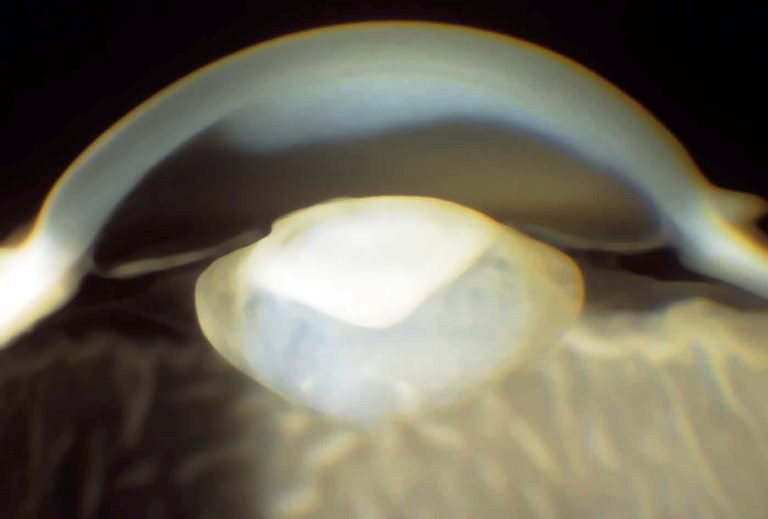
Se la cataratta è monolaterale, oltre all’ambliopia si sviluppa generalmente strabismo mentre in quella bilaterale evoluta un segno tipico è il nistagmo che insorge intorno ai 3 mesi di età.
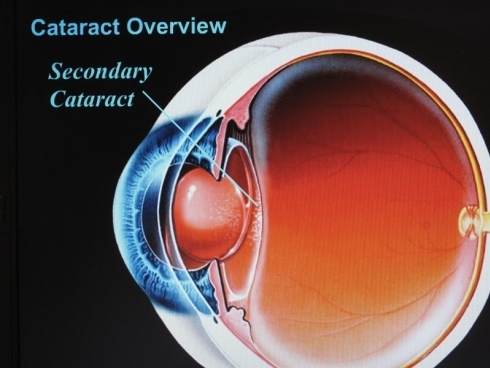
Si manifesta nel 10 per cento degli operati di cataratta ed è un processo di opacizzazione che riguarda non più il cristallino, ma la capsula in cui è stato inserito. I sintomi sono simili a quelli della cataratta primaria: visione sfocata, perdita progressiva della capacità visiva.
L’opacizzazione della capsula è dovuta alla proliferazione di cellule infiammatorie che, per motivi non ancora del tutto chiariti, depositandosi su di essa la rendono opaca. E’ certo che la cataratta secondaria colpisce maggiormente i giovani e le persone soggette ad uveiti od altre infiammazioni croniche.
E’ la cavità oculare priva del bulbo e consiste nell’orbita con i suoi contenuti (periosteo, grasso orbitale, sacco congiuntivale, capsula di Tenone, residui dei muscoli oculomotori).
La cavità anoftalmica può essere di origine congenita a causa del mancato o incompleto sviluppo del bulbo oculare, oppure chirurgica in seguito alla sua rimozione.

Secondo l’OMS un soggetto è cieco quando l’acuità visiva corretta nell’occhio migliore è inferiore a 1/20, mentre è ipovedente quando essa è compresa tra 3/10 e 1/20. In dettaglio sono state definite cinque categorie:
Le altre tre categorie riguardano, invece, il soggetto cieco:
3° cat. = visus 1/20-1/100
4° cat. = visus 1/100-P.L.
5° cat. = visus spento
In Italia il concetto legale di cecità-ipovisione è stato ridefinito con la legge 3 aprile 2001, n. 138 che prende in esame, per la valutazione del danno, non solo la visione centrale ma anche quella periferica (campo visivo).
La precedente legge (n. 382/70), invece, quantificava la menomazione visiva sulla base della sola acuità visiva: accadeva così che un paziente affetto da retinite pigmentosa o glaucoma avanzato e con un campo visivo ridotto a meno di 5-10 gradi, non fosse nemmeno riconosciuto come ipovedente.
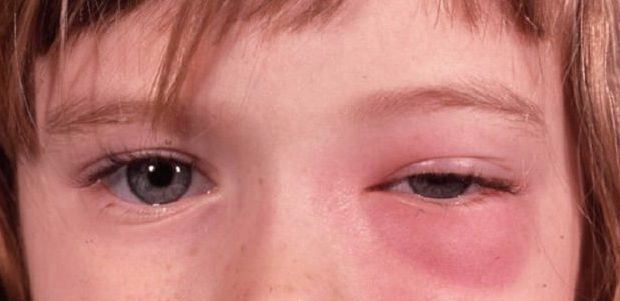
Infezione caratteristica dell’infanzia che causa tumefazione acuta e arrossamento della palpebra e della cute circostante (cellulite periorbitale) o anche dei tessuti orbitali (cellulite orbitale) che può essere causata da un focolaio d’infezione esogeno (ferita o puntura d’insetto) o endogeno.
L’infezione può anche essere secondaria, causata da una primitiva infezione dei seni etmoidali o da infezioni cutanee locali e può diffondersi in entrambe le direzioni attraverso le vene oftalmiche inferiore e superiore.
Un terzo dei pazienti ha comunque una storia di trauma od infezione locale.
Il primo sintomo è la tumefazione e l’arrossamento della palpebra (quasi sempre monolaterale) ma spesso la proptosi spinge il bulbo verso l’esterno, lateralmente o in basso.
Le complicanze più frequenti sono la trombosi dell’arteria centrale o della vena retiniche e un danno retinico secondario causato dall’aumentata pressione intraoculare. Quando l’infezione non rimane circoscritta all’orbita, le possibili complicanze endocraniche sono gli ascessi epidurali, subdurali o cerebrali, la trombosi del seno cavernoso o della vena corticale e la meningite batterica. I bambini affetti da cellulite orbitaria necessitano di ospedalizzazione e di trattamento tempestivo.
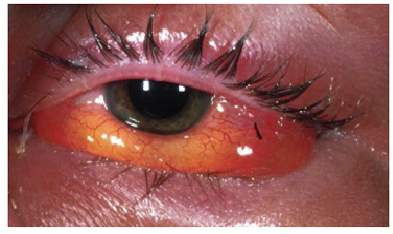
Edema congiuntivale causato da un’infiltrazione sierosa spesso dovuto ad una reazione allergica. La congiuntiva così rigonfia, assume un colorito roseo o biancastro e può addirittura arrivare a sporgere dalla palpebra.
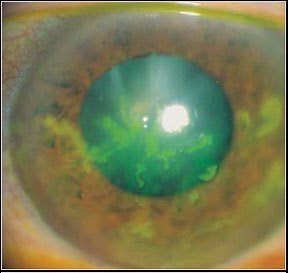
Cheratite erpetica
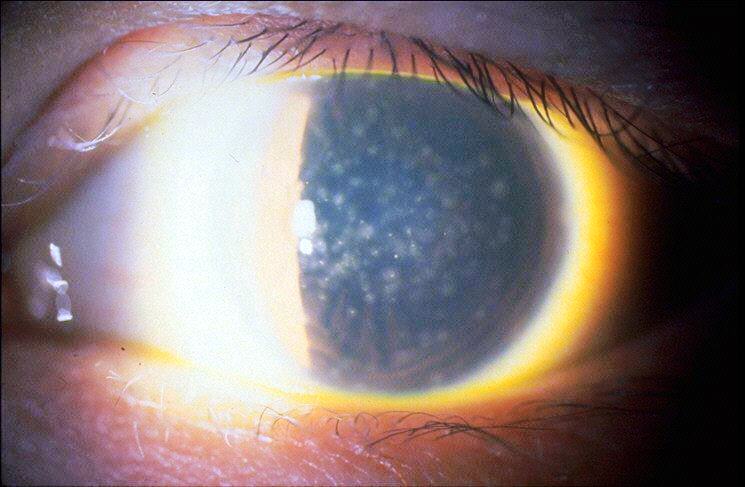
Cheratite Puntata
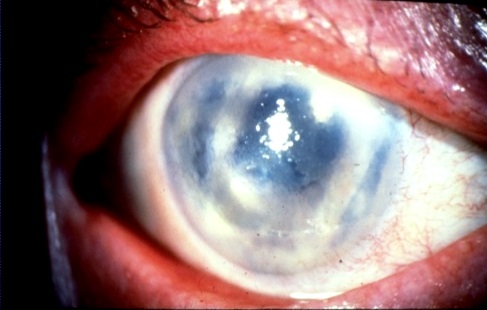
Cheratite bollosa
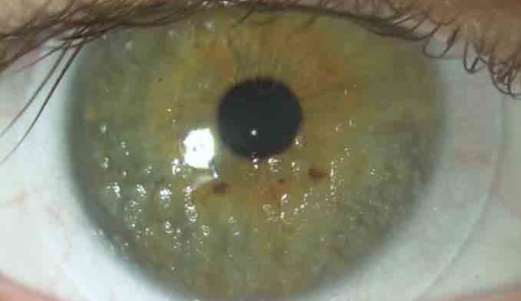
Cheratite superficiale
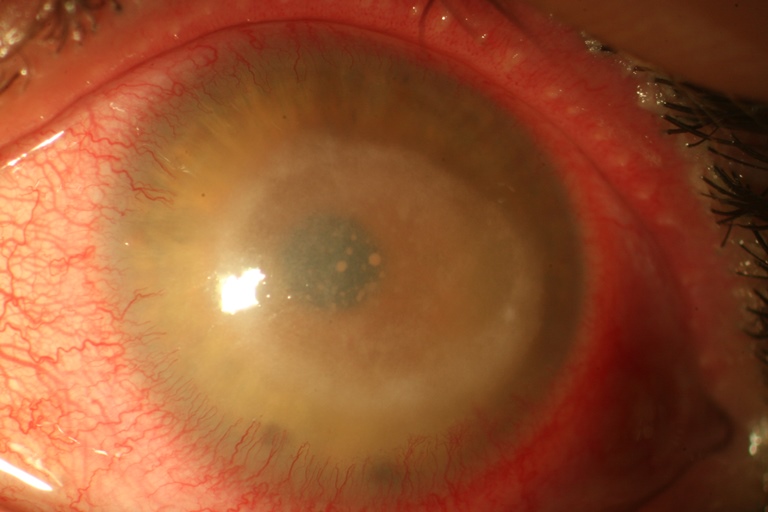
Cheratite da Acanthamoeba
Patologia infiammatoria o infettiva della cornea che può essere causata da fattori infettivi (virus, batteri, protozoi, funghi), fisici (raggi ultravioletti) o malattie sistemiche (artriti reumatoidi o vasculiti disseminate). Se non opportunamente curata, può causare la distruzione più o meno vasta e profonda dello strato corneale.
I sintomi tipici sono annebbiamento o calo della vista, dolore, fotofobia e si manifestano con piccole erosioni superficiali dell’epitelio e opacità disseminate all’interno della cornea (infiltrati stromali). Altra caratteristica è la crescita dei vasi sanguigni all’interno del tessuto corneale a partire dal limbus con formazione di tessuto fibroso sotto l’epitelio (panno corneale). Questa patologia è spesso contraddistinta da edema corneale, cioè da un eccesso di liquido all’interno della cornea.
Le forme infettive presentano spesso dolore, secrezione purulenta e torbidità dell’umore acqueo. In relazione alla profondità dell’ulcerazione la cheratite può essere superficiale o perforata.
I pazienti più a rischio sono:
La cheratite è classificata in:
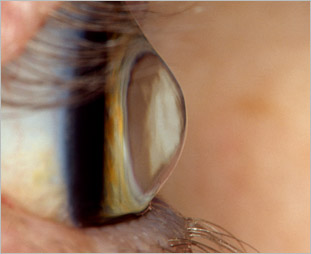
Patologia della cornea che consiste nel suo progressivo sfiancamento del tessuto che si assottiglia e prolassa all’apice assumendo la forma di un cono (ectasia). In genere colpisce un abitante ogni 1.500 ed è bilaterale ma i casi di alterazioni riconducibili al cheratocono sono almeno il doppio. L’esordio e l’evoluzione della malattia sono molto variabili e negli stadi iniziali è possibile correggere il difetto con occhiali ma con la sua evoluzione è possibile solo l’applicazione di lenti a contatto speciali. Negli stadi avanzati, il 10% del totale, quando l’assottigliamento estremo del tessuto corneale comporta un rischio imminente di perforazione, la cheratoplastica è l’unica soluzione. Poiché il tessuto corneale è privo di vasi sanguigni il rigetto è meno frequente che nel trapianto di altri organi.
Le cause del cheratocono sono sconosciute ma sembra che il fattore genetico sia il più probabile, oltre a fattori esterni come microtraumi da sfregamento o da allergie che ne condizionano l’evoluzione. L’aumento di alcuni enzimi specifici, fra cui le proteasi e una diminuzione dei loro inibitori, determina un difetto di funzionalità dei cheratociti con conseguente riduzione dello spessore e degenerazione e deformità della cornea.
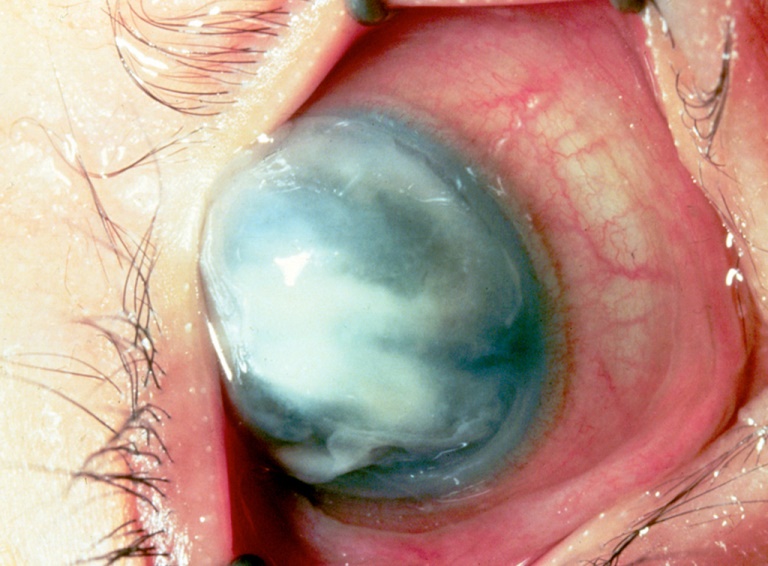
Patologia associata a carenza di vitamina A e malnutrizione calorico-proteica, caratterizzata da una cornea secca e torbida che si sfalda. Può essere l’estrema complicanza della Xeroftalmia.
L’ulcerazione corneale con infezione secondaria è una diretta conseguenza ed anche le ghiandole lacrimali e la congiuntiva sono interessate. La mancata produzione di lacrime causa una estrema secchezza degli occhi, mentre nella congiuntiva bulbare compaiono chiazze spugnose di colore grigio-biancastro. Può essere associata a cecità notturna.
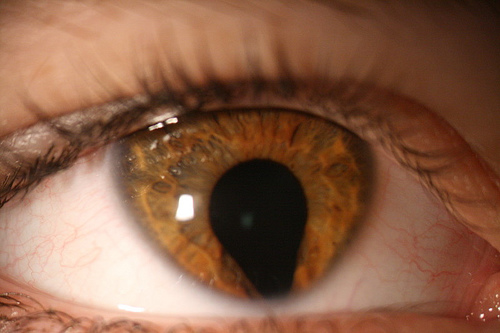
Anomalia malformativa congenita dell’occhio caratterizzata dalla presenza di una fessurazione che interessa le strutture della parte inferiore del bulbo oculare per lo più ne è colpita l’iride, per cui la pupilla non è più rotonda ma ha forma di goccia. La lesione può essere isolata (casuale e non associata ad altre patologie) o in rari casi sindromica, cioè estesa anche al corpo ciliare, alla coroide, al cristallino, alla retina, al nervo ottico, ed associata ad altre anomalie malformative dell’occhio o di altri organi (cuore, sistema nervoso centrale) nel quadro di sindromi di tipo sistemico come ad esempio la Sindrome di Charge.
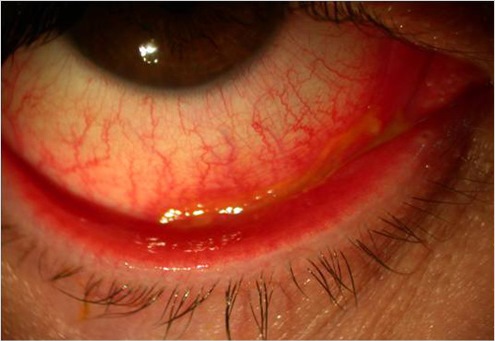
Congiuntivite acuta
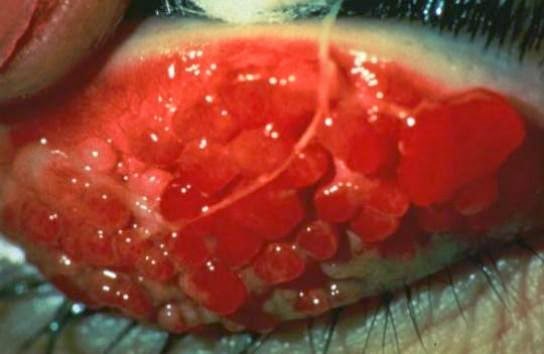
Congiuntivite giganto-papillare
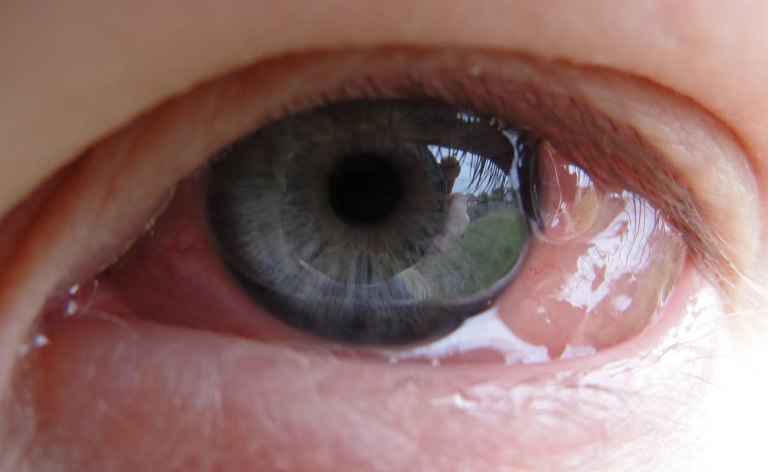
Congiuntivite allergica
Flogosi della congiuntiva che può essere di natura infiammatoria, allergica o infettiva (virale o batterica). E’ dovuta all’azione di germi patogeni (batteri, virus, miceti e protozoi) o sostanze con potere allergizzante (polvere, farmaci, ecc..) o sostanze tossiche. I sintomi sono differenti e variano in base alla sua origine:
– Il bulbo è dolente, con sensazione di bruciore e di corpo estraneo o «sabbia negli occhi».
– La congiuntiva è iperemica, edematosa, simile ad una gelatina traslucida.
– La lacrimazione e la secrezione sono abbondanti.
La congiuntivite è classificata in:
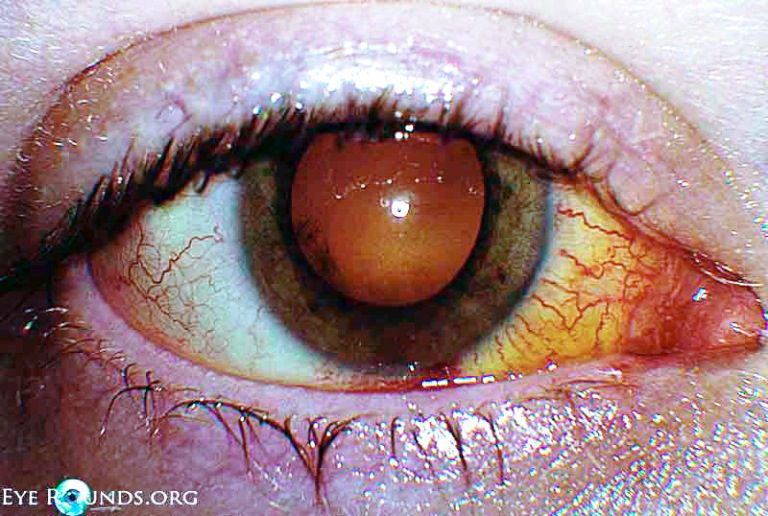
La corioretinite infettiva (da batteri, virus, miceti, parassiti) può essere provocate da toxoplasmosi, toxocariasi, sifilide, infezioni erpetiche, candidosi e rosolia. Nei pazienti con immunodepressione primaria autoimmune o acquisita (AIDS) è frequente la corioretinite da citomegalovirus, criptococco e histoplasma.
Le complicanze, anche invalidanti per la funzione visiva, sono l’edema papillare del nervo ottico, la maculopatia cistoide, il distacco di retina e l’endoftalmite.
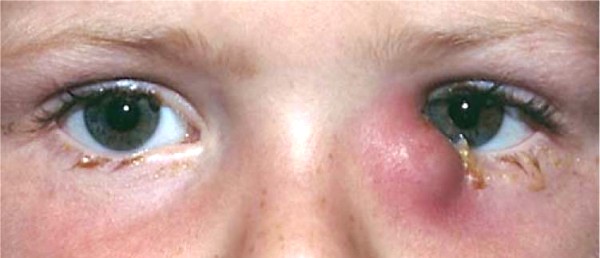
Infiammazione del sacco lacrimale. Nella fase acuta si presenta con la comparsa di una massa arrossata e dolente all’altezza del canto interno e con abbondante epifora, poiché i canali lacrimali ostruiti non riescono a drenare le lacrime nella fossa nasale. Se la condizione infiammatoria permane può mutare in una dacriocistite cronica con un’ostruzione completa del sacco lacrimale senza possibilità di passaggio per le lacrime. In questo caso, se la terapia antibiotica non è sufficiente, occorre intervenire chirurgicamente ripristinando la pervietà delle vie lacrimali.
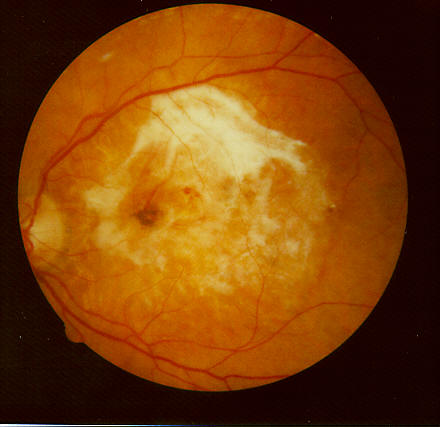
La degenerazione maculare senile è una delle cause più frequenti di cecità senile nei Paesi Occidentali e consiste in una malattia cronica distrofico-degenerativa che colpisce la macula, regione centrale della retina, responsabile della visione definita. L’evoluzione di tale malattia è progressiva e col tempo tende a compromettere la visione in modo sempre più grave. Esistono due forme di DMS: Nella DMS «secca» compaiono lesioni caratteristiche denominate Drusen, accumuli di scorie cellulari che possono riassorbirsi o calcificare.
Nella DMS «umida» oltre alle Drusen si formano dei neovasi sotto la retina (membrana neovascolare sottoretinica) responsabili dell’evoluzione essudativa della DMS. Nelle fasi iniziali può non dare sintomi, soprattutto se è monolaterale. Si può notare riduzione della visione centrale, sfuocamento delle parole nella lettura, un’area scura al centro del campo visivo (scotoma) e distorsione delle linee dritte (metamorfopsie). Questo sintomo deve indurre ad una visita oculistica urgente.
Nella forma “secca” agli accumuli di scorie possono seguire alterazioni atrofiche della retina con conseguente calo della vista. In quella “umida” i neovasi e le emorragie sottoretiniche alterano la funzione visiva centrale.
Nella DMS «umida» oltre alle Drusen si formano dei neovasi sotto la retina (membrana neovascolare sottoretinica) responsabili dell’evoluzione essudativa della DMS. Nelle fasi iniziali può non dare sintomi, soprattutto se è monolaterale. Si può notare riduzione della visione centrale, sfuocamento delle parole nella lettura, un’area scura al centro del campo visivo (scotoma) e distorsione delle linee dritte (metamorfopsie). Questo sintomo deve indurre ad una visita oculistica urgente.
Nella forma “secca” agli accumuli di scorie possono seguire alterazioni atrofiche della retina con conseguente calo della vista. In quella “umida” i neovasi e le emorragie sottoretiniche alterano la funzione visiva centrale.
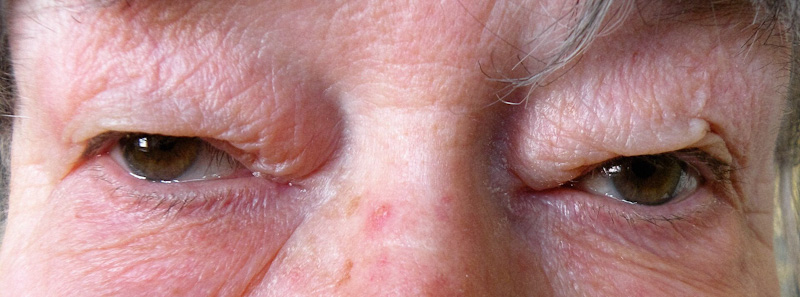
La dermatocalasi è una condizione tipica dell’età avanzata, caratterizzata da eccesso di cute nella palpebra superiore, che tende a scendere progressivamente fino ad interferire con il campo visivo.
Questa condizione è spesso associata alla ptosi della palpebra e a prolasso del grasso orbitario e se la cute discendente coinvolge le ciglia, può provocare un entropion. Inoltre, la cute più lassa è più delicata ed è soggetta ad irritazione e gonfiore. Questa condizione può essere causata da alcune patologie tiroidee o l’insufficienza renale, così come i traumi e le patologie del tessuto connettivo. La predisposizione genetica ed il fumo possono aggravare tale condizione.
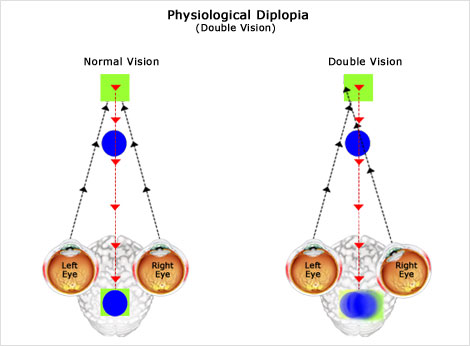
Alterazione della funzione visiva che consiste nella visione sdoppiata delle immagini, dovuta alla percezione di due immagini distinte a livello dei centri ottici superiori (chiasma, mesencefalo). Nell’adulto la causa più frequente è la paralisi dei muscoli estrinseci che fanno mancare il parallelismo tra gli assi retinici e di conseguenza la formazione di due immagini che non riescono ad essere sovrapposte l’una all’altra.
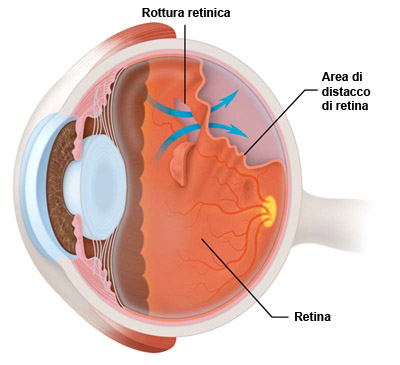
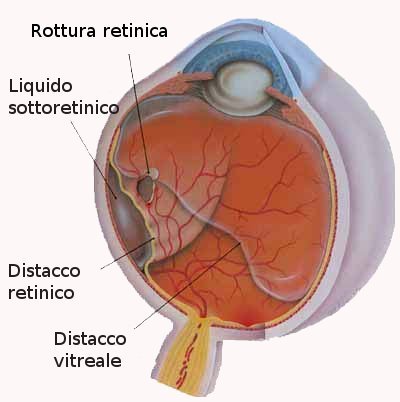
La retina viene mantenuta nella sua sede dalla coroide e dal corpo vitreo. Ogni strappo o foro nella retina provoca una fuoriuscita del vitreo che si insinua tra retina e coroide, separandole. Ciò si verifica anche quando uno strato della retina si solleva trascinando con sé i vasi sanguigni che la alimentano. Dopo 48 ore dal distacco inizia la morte delle cellule e la perdita progressiva della vista, specie se la porzione di retina distaccata è quella centrale. Il DDR può essere:
Si manifesta con la comparsa di corpi scuri fluttuanti (miodesopsie) uno scotoma in un settore del campo visivo, flash luminosi persistenti (fotopsie) e improvviso calo del visus.
Patologia genetica non infiammatoria. Può essere anche di tipo acquisito o sistemico, associato cioè ad una sindrome preesistente. Si manifesta con opacità corneali di varia forma che possono causare un deficit visivo. In base alla profondità della lesione si dividono in:
Una delle più comuni distrofie acquisite (o degenerazione corneale) è il gerontoxon che è un deposito di lipidi a livello del limbus che si manifesta in età senile, assottigliando la cornea nella zona perilimbare (arco senile).
Le distrofie corneali gravi che compromettono la visione sono trattate chirurgicamente con cheratoplastica lamellare
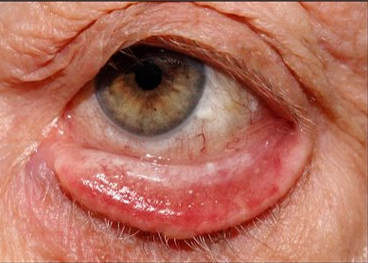
Malposizione del margine palpebrale superiore (molto raro) o inferiore, che si manifesta con l’eversione permanente dello stesso e può essere senile, paralitico, cicatriziale o meccanico. Per tutti i casi di origine non dermatologica la terapia è essenzialmente chirurgica.
ECTROPION SENILE
È causato da una progressiva lassità del tarso e dei legamenti cantali, la palpebra inferiore diviene lassa e non ha più la stabilità meccanica che ne assicura funzione e posizione.
ECTROPION PARALITICO
E’ causato dalla paralisi del nervo facciale e si associa alla retrazione della palpebra superiore e ptosi sopraccigliare.
ECTROPION CICATRIZIALE
È causato per lo più a lesioni traumatiche della cute palpebrale, come ferite, cicatrici, ustioni o causticazioni oppure pregresse terapie chirurgiche.
ECTROPION MECCANICO
Può essere causato da neoformazioni del bordo palpebrale che determinano lo spostamento del margine palpebrale verso l’esterno a causa del loro volume.
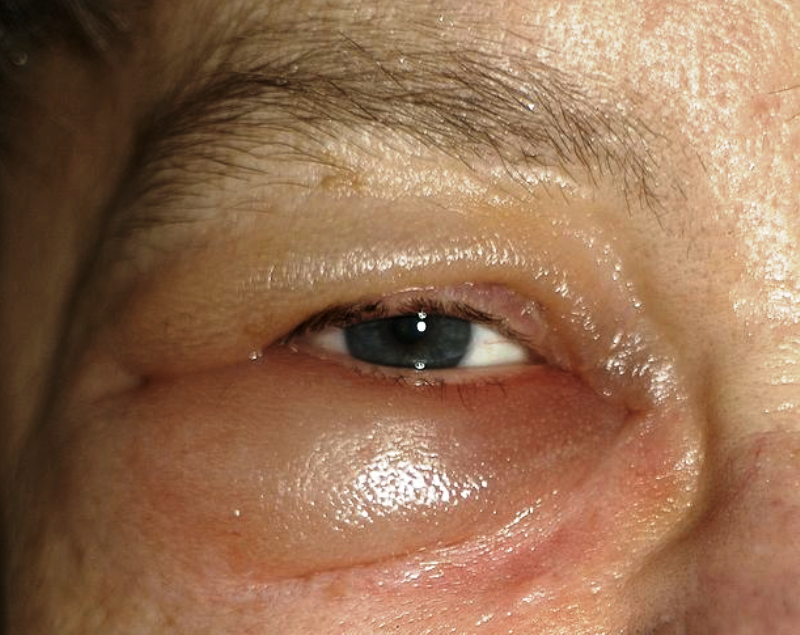
L’edema oculare si riferisce alla presenza di liquidi in eccesso nei tessuti connettivi intorno all’occhio. Può essere causato da traumi del bulbo, infezioni, lesioni o dai processi cicatriziali post-chirurgici.
Si manifesta con gonfiore e forte arrossamento, dura alcuni giorni e si cura con terapia antinfiammatoria. E’ riscontrabile nei tessuti suturati dopo un intervento ed ancora in fase cicatriziale.
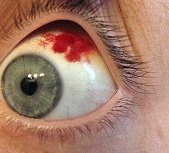
Emorragia sottocongiuntivale
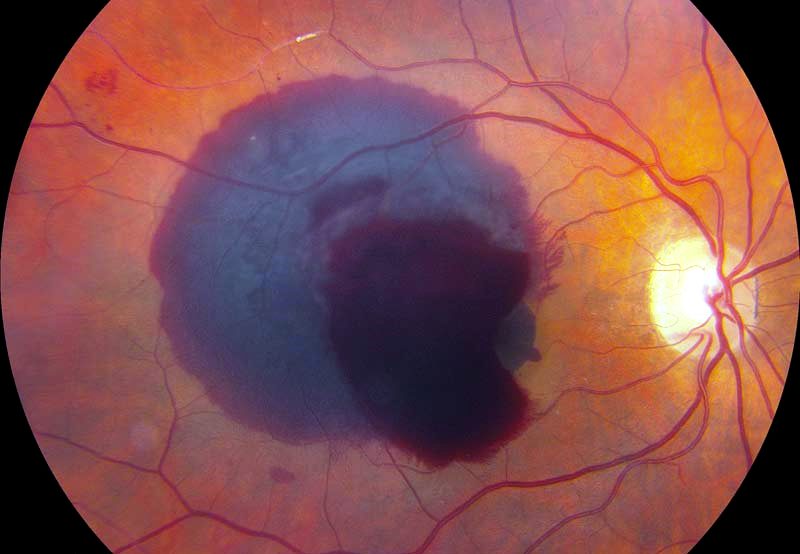
Emorragia retinica
Le Emorragie sottocongiuntivali possono prodursi in seguito a piccoli traumi, sforzi, starnuti o colpi di tosse. Non sono sintomo di alcuna patologia, si presentano come fuoriuscita di sangue al di sotto della congiuntiva e vengono riassorbite spontaneamente in genere entro un paio di settimane.
Le emorragie vitreali (emovitreo) possono verificarsi in seguito all’occlusione venosa retinica, alla retinopatia diabetica, al distacco posteriore del vitreo, alla neovascolarizzazione e la rottura retiniche o ai traumi oculari.
Possono causare un distacco di retina e tendono a regredire molto lentamente.
Le emorragie retiniche sono a forma di fiamma nello strato più superficiale della retina, come avviene in caso di ipertensione o di occlusione venosa, oppure possono essere tondeggianti negli strati retinici più profondi, come nel diabete mellito o negli infarti settici.
Le emorragie retiniche sono sempre indicative di una malattia vascolare generalmente sistemica.
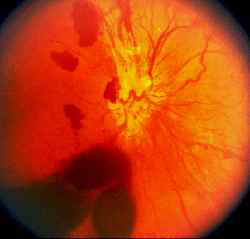
E’ il versamento ematico causato da un’emorragia che invade la camera posteriore dell’occhio dal corpo vitreo. L’emorragia può originare dalla rottura di un vaso retinico in corso di un distacco di vitreo con conseguente lacerazione retinica o dalla rottura spontanea di neovasi nelle retinopatie di tipo ischemico quali la retinopatia diabetica proliferante o negli esiti di una occlusione venosa retinica. E’ causato frequentemente anche da traumi contusivi o DMS emorragica.
Il sintomo è un calo repentino del visus con sensazione di nebbia che cresce velocemente fino al completo oscuramento della visuale (che comunque rimane luminosa contrariamente al distacco della retina).
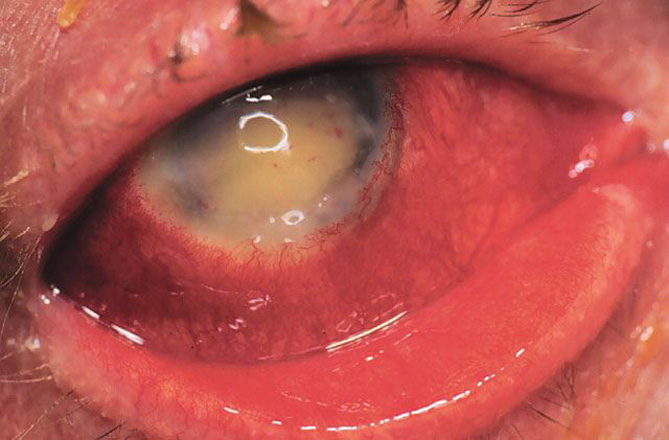
Grave infezione endoculare massiva che può coinvolgere il vitreo, la retina, l’uvea e la sclera ed evolvere poi in panoftalmite (processo infiammatorio diffuso che colpisce tutte le strutture del bulbo oculare). Si tratta di una delle più gravi emergenze oculistiche: l’infezione può estendersi rapidamente oltre il bulbo colpendo la cavità orbitaria ed il sistema nervoso centrale.
Nella sua forma primaria segue spesso una ferita perforante del bulbo se a questa sopravviene un’infezione. Nella forma secondaria può derivare da un’infezione tardiva anche in bulbi apparentemente guariti dopo un intervento. Può causare fenomeni simili nell’altro occhio per “simpatia”.
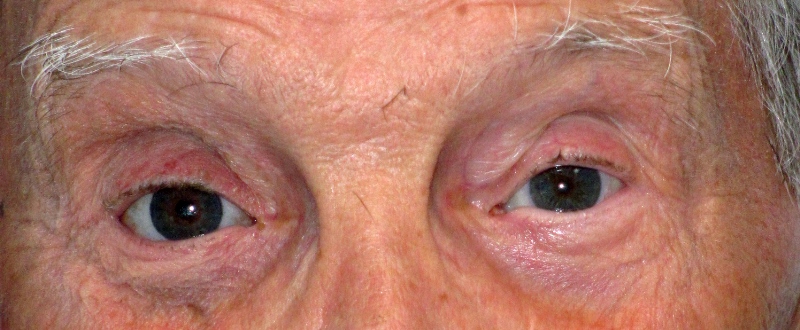
Condizione in cui il bulbo è infossato nella cavità orbitaria. Può manifestarsi dopo la frattura del pavimento orbitario o nella sindrome della cavità anoftalmica in cui si associa all’aumento del solco palpebrale superiore, alla pseudoptosi della palpebra superiore ed alla lassità della palpebra inferiore. E’ il maggiore e più diffuso difetto morfologico nei pazienti che hanno subito enucleazione del bulbo a causa del riassorbimento fisiologico del grasso endorbitario dopo l’intervento.
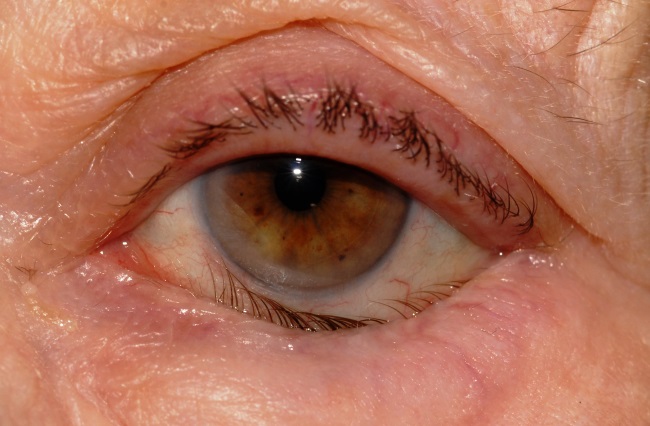
Rivolgimento verso l’interno del margine libero della palpebra (prevalentemente quella inferiore) e delle ciglia che tendono a crescere verso l’interno e può interessare la palpebra superiore o quella inferiore. L’attrito delle ciglia sulla cornea può causare dolore, arrossamento, sensazione di corpo estraneo e lacrimazione, fino a produrre abrasioni o ulcere corneali. La terapia è essenzialmente chirurgica.
Esistono tre tipi di entropion: spastico, involutivo e cicatriziale e può essere associato a modificazioni secondarie: deviazione della direzione delle ciglia (trichiasi) e sviluppo di ciglia aberranti. Inoltre può essere l’esito cicatriziale di ustioni chimiche, di traumi, di un tracoma o di un pemfigoide oculare.
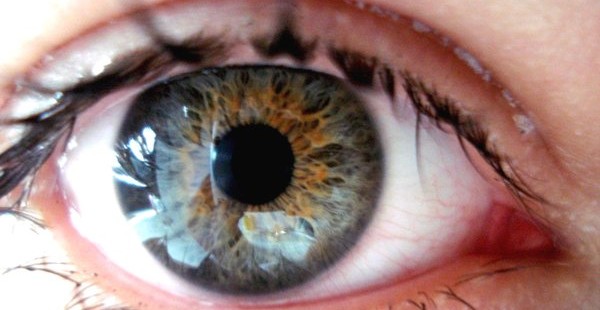
Flusso lacrimale patologico che può essere conseguente al restringimento delle vie lacrimali di deflusso, ovvero di quel sistema di condotti che raccolgono le lacrime dal sacco congiuntivale e le convogliano verso le cavità nasali.
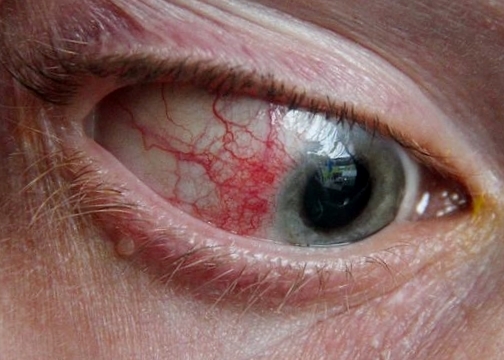
L’episclerite è una patologia infiammatoria che interessa gli strati superficiali della sclera. I sintomi dell’episclerite includono lieve dolore agli occhi, arrossamento e lacrimazione, mentre la capacità visiva non è influenzata. I pazienti con episclerite lamentano fotofobia meno marcata rispetto ai pazienti affetti da uveite.
Esistono due tipi di episclerite: il tipo nodulare ed il tipo foruncoliforme.
L’episclerite nodulare si presenta come un nodulo che solleva la congiuntiva bulbare in un punto con arrossamento limitato ad una piccola zona, dolore lieve e nessun problema al visus. Colpisce maggiormente gli adulti e gli anziani con predisposizione ai disturbi reumatici o che soffrono di iperuricemia.
Nell’episclerite foruncoliforme metastatica che può manifestarsi a qualsiasi età, si nota un’area iperemica attorno al limbus con formazione di un piccolo rialzamento arrossato della dimensione di un pisello o anche più piccolo che in breve tempo cambia colore, diventando giallastro. Dopo circa otto giorni si apre spontaneamente attraverso la congiuntiva con fuoriuscita di secrezione giallastra e formazione di un lembo necrotico che indica la sua definitiva guarigione.
Protrusione (spostamento in avanti) del globo oculare rispetto alla sua sede naturale causato dalla spinta proveniente dall’interno dell’orbita.
Il lieve esoftalmo non è sintomatico ma i sintomi si manifestano quando la protrusione in avanti è tale da disallineare i due assi visivi dando origine a diplopia. La dislocazione dell’occhio può essere tale per cui le palpebre non siano abbastanza lunghe per coprire l’occhio, determinando una cheratite da esposizione.
Le cause sono tutte le alterazioni che creano un aumento di volume del contenuto orbitario. Si possono classificare in tumorali (crescita di una massa nella cavità orbitaria), infiammatorie (l’edema dato dall’infiammazione aumenta il volume dei tessuti) e l’infiammazione può essere dovuta a cause fisiche, infettive o traumatiche e anche ad alterazioni vascolari come la fistola carotido-cavernosa.
Tra le cause più frequenti c’è anche il morbo di Basedow la cui causa è dovuta all’ipertiroidismo, per cui si ha un aumento del volume dei muscoli dell’occhio a causa dell’edema e del grasso retrobitario che genera un esoftalmo in genere bilaterale, oltre ad altri segni caratteristici della patologia come l’alterata motilità palpebrale al movimento degli occhi.
Liquido ricco di proteine e polisaccaridi ma anche di linfociti, globuli rossi e residui cellulari che si forma all’interno del corpo vitreo. La formazione dell’essudato è conseguenza di un processo infiammatorio (forme essudative) che possono essere sintomi di distacco di retina essudativo, retinopatia diabetica, uveite o anche di forme neoplastiche sottoretiniche. In questo caso, dato che gli essudati duri sono sempre il risultato di una fuoriuscita di liquido dai capillari nello spessore della retina, si associano di solito a edema retinico. Gli essudati cotonosi sono invece zone biancastre della retina, dall’aspetto di fiocchi di cotone. Sono provocate dall’interruzione del flusso di ossigeno e di sostanze nutritizie dovuto dall’occlusione di capillari dalla parete troppo spessa
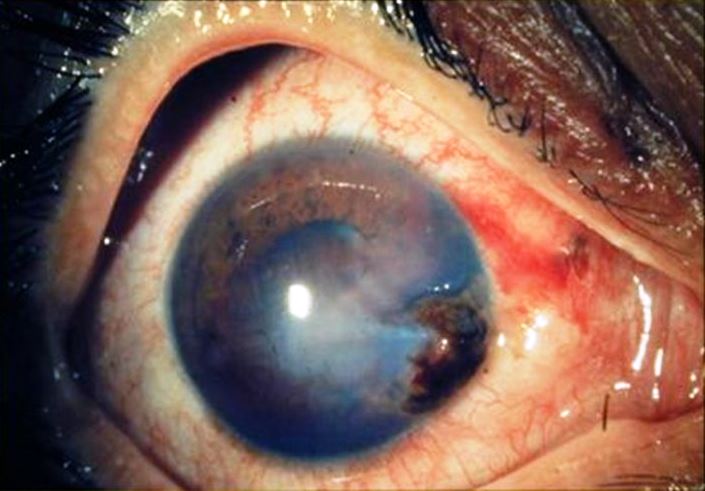
Ha sede generalmente nell’emisfero anteriore del bulbo e sono prodotte frequentemente da oggetti appuntiti o taglienti. Qualsiasi sia il tipo di lesione queste ferite sono quasi sempre gravi, con interessamento dell’iride e del cristallino quando non addirittura della camera posteriore e delle strutture retiniche. Si dividono in:
Ferita corneale semplice, spesso non si limita ad attraversare la cornea ma produce le lesioni al cristallino e all’iride, provocando spesso la subatrofia del bulbo
Ferita della congiuntiva e della sclera, le piccole ferite congiuntivali senza ritenzione di corpi estranei guariscono spesso rapidamente mentre a volte il trauma può interessare la congiuntiva e la sclera, con il rischio di versamento di sangue nel vitreo e necessità di intervenire chirurgicamente
Ferita con presenza di corpi estranei, se il corpo estraneo attraversando la cornea raggiunge l’iride e il cristallino può procurare un’emorragia endobulbare con complicanze anche molto gravi: i corpi estranei in vetro e alluminio sono i meno pericolosi mentre il ferro e il rame spesso provocano siderosi e calcolosi del bulbo.
Altre complicanze tipiche sono le sinechie, la cataratta traumatica, il distacco di retina che possono portare alla ftisi del bulbo.

ipersensibilità oculare alla luce, accompagnata da lacrimazione ed a volte associata a congiuntiviti, cheratiti o altre patologie come l’Herpes Simplex.
Può essere un disturbo temporaneo dopo un intervento chirurgico oppure dovuto ad un uso improprio delle lenti a contatto o ancora, può essere l’effetto collaterale dell’assunzione di farmaci.
Fenomeni avvertiti come lampi di luce o flash luminosi a volte anche colorati, visibili dal paziente in presenza di alterazioni retiniche o trazioni vitreali. Possono essere causati da alterazioni periferiche come fori retinici o da patologie più gravi come il distacco di retina o da un iniziale distacco posteriore del corpo vitreo. Raramente si possono presentare in chi non ha alcuna patologia oculare.
Le fratture del distretto maxillo-facciale interessano diverse ossa dello scheletro facciale. Le fratture più frequenti sono quelle delle ossa nasali, quelle del complesso orbito-maxillo-zigomatico con possibile coinvolgimento del pavimento orbitario (blow out) e le fratture della mandibola. Le fratture delle pareti orbitarie possono interessare frequentemente zigomo, naso ed etmoide.
In base alla loro posizione le fratture orbitali si dividono in fratture del pavimento orbitale, della parete mediale, di quella laterale e del tetto dell’orbita.
In base alla loro natura patogenetica le fratture orbitali possono essere di tipo blow-out (scoppio esterno all’orbita) o blow-in (scoppio interno all’orbita). I segni tipici della frattura orbitale sono l’edema e l’ematoma periorbitario con interessamento congiuntivale, l’enoftalmo e la diplopia, soprattutto nello sguardo verso l’alto. In alcuni casi la muscolatura estrinseca dell’occhio che determina i movimenti del bulbo oculare può rimanere imprigionata nella breccia della frattura determinando un deficit di motilità oculare che se non prontamente corretto può divenire permanente, a causa del processo fibrotico della fase cicatriziale.
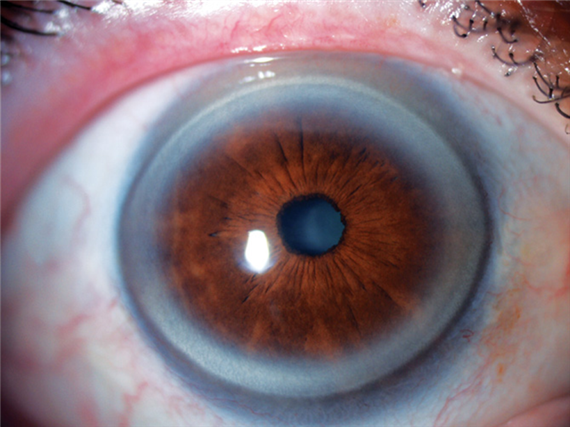
Processo degenerativo della cornea (distrofia) che si presenta sotto forma di un’aureola opaca biancastra perilimbare del diametro di 2-3 mm. Si tratta di un accumulo di depositi lipidici localizzato a livello del limbus che si manifesta progressivamente in età senile, assottigliando la cornea nella zona corrispondente.
Non arreca alcun danno alla visione e non necessita di alcun trattamento.
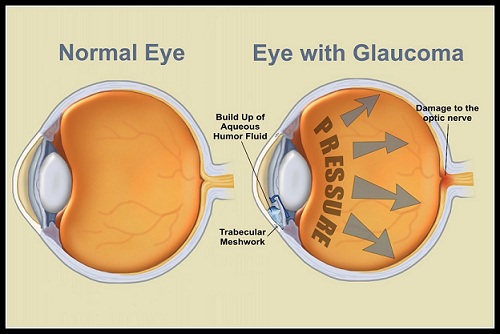
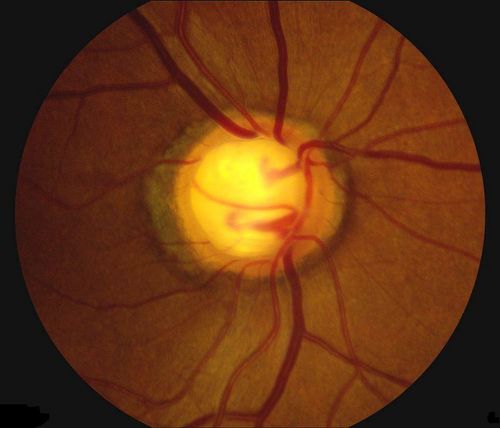
Patologia il cui principale fattore di rischio è un’elevata pressione intraoculare (IOP). Altri fattori di rischio sono l’età (sopra i 40 anni), la familiarità per la malattia, l’etnia nera, la miopia superiore a 4 diottrie ed altre patologie vascolari. La pressione interna (tono) aumenta perché l’umore acqueo prodotto in continuazione all’interno del bulbo incontra difficoltà a defluire verso l’esterno attraverso il trabecolato ed i canali di Schlemm.
I dati indicano tra 10 e 21 mm Hg i valori “normali” di pressione intraoculare. Il tono elevato provoca una pressione continuata sul nervo ottico che se non curato causa una riduzione permanente della vista, ma oggi le terapie mediche e chirurgiche consentono un buon controllo della malattia, soprattutto se la diagnosi viene fatta precocemente.
Il Glaucoma è classificato in:
Glaucoma primario ad angolo aperto (POAG) inclusi ipertensione oculare, quello a tensione normale e quello giovanile. Si tratta della forma più frequente (80% dei casi) e si verifica in seguito a un ostacolo che incontra l’umor acqueo nel defluire attraverso il trabecolato. Si sviluppa lentamente ed è spesso asintomatico, per cui si può rilevare la presenza della patologia solo quando il danno al nervo ottico è già avanzato.
La patologia glaucomatosa provoca una perdita irreversibile delle strutture nervose e della funzione visiva. All’esordio i sintomi soggettivi sono praticamente assenti; il paziente può accorgersi di una limitazione del campo visivo urtando gli ostacoli o inciampando negli scalini, quando ormai il danno è molto avanzato. Per questa ragione la diagnosi precoce rappresenta il punto nodale nella cura del glaucoma.
Il glaucoma acuto è stato in passato ed in parte è ancora una delle prime cause di enucleazione del bulbo oculare.
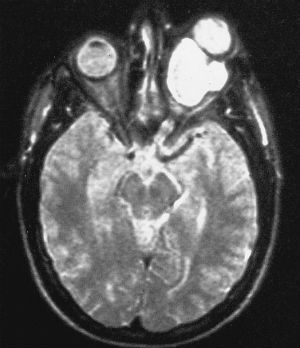
E’ una forma rara di tumore cerebrale a crescita lenta, riscontrabile nei bambini che inizia dalla parte intraorbitaria del nervo ed in rari casi tende ad estendersi verso il chiasma ma senza infiltrare i tessuti vicini.
L’esame neurologico evidenzia generalmente alterazione dei nervi ottici e può essere presente anche l’aumento della pressione endocranica.
Il glioma si manifesta con rapido calo del visus, proptosi ed esoftalmo progressivo ed irriducibile. All’oftalmoscopio la papilla può evolvere verso l’atrofia ottica. Il trattamento è solo chirurgico, con una pessima prognosi per la funzione visiva ma buona per l’aspettativa di vita se l’intervento è precoce, poiché il tumore ha bassa aggressività e le recidive sono rare.
Il trattamento del glioma può provocare effetti collaterali a lungo termine. Colpendo generalmente i bambini, gli effetti della radioterapia o della chemioterapia potrebbero non palesarsi immediatamente e possono causare difficoltà cognitive, disturbi dell’apprendimento e della crescita.
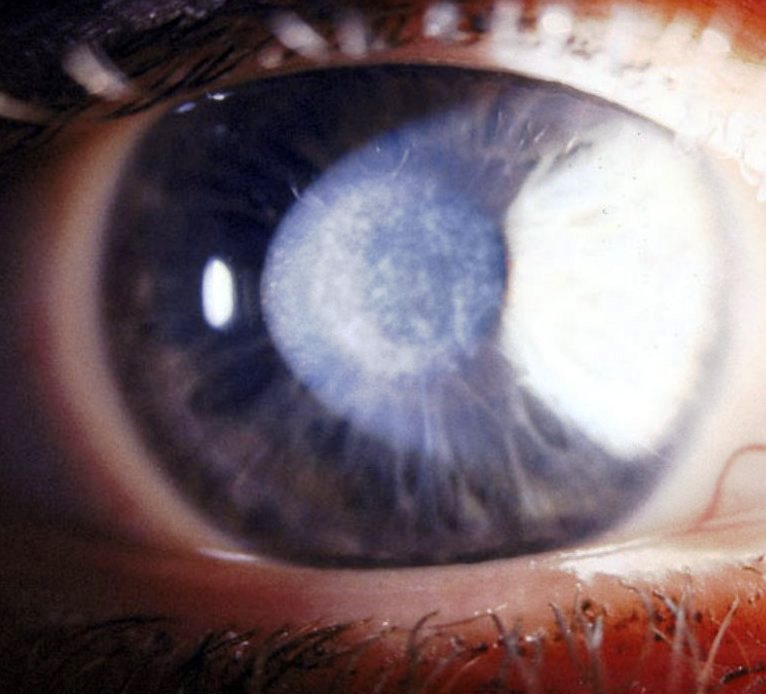
Cicatrice corneale di origine fibrotica che può presentarsi come complicanza dopo chirurgia refrattiva. Può avere vario grado di invasività ed influenzare la qualità della visione post-operatoria, a volte peggiorandola, dato che il trattamento interessa proprio la parte centrale della cornea, cioè quella deputata alla visione.
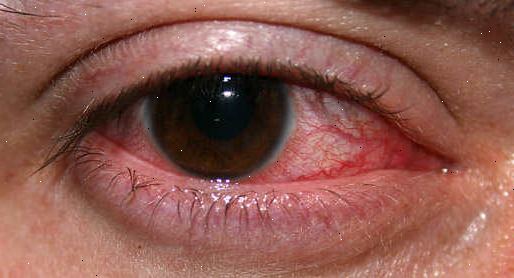
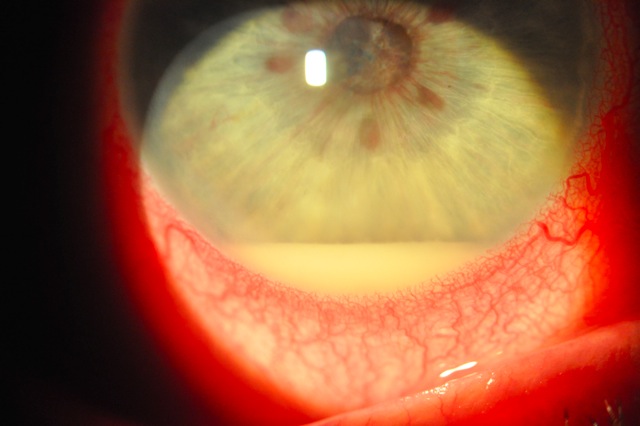
Il virus dell’Herpes Simplex (HSV) è responsabile di un ampio spettro di malattie. L’HSV è un virus pandemico e l’uomo rappresenta l’unico serbatoio del virus. Le manifestazioni oculari dell’infezione da HSV possono interessare tutti i distretti dell’occhio e la cheratite è la patologia più comune.
Nella congiuntivite erpetica, nei due terzi dei casi, dopo circa due settimane si ha interessamento della cornea con fotofobia, dolore e riduzione dell’acuità visiva. Essa può manifestarsi con una diffusa cheratite punteggiata epiteliale o una cheratite dendritica fino alla comparsa di una cheratite disciforme. Nella maggior parte dei casi le lesioni guariscono senza esiti. Dopo l’infezione primaria il virus rimane allo stato latente nel sistema nervoso per poi, in particolari casi, riattivarsi con frequenza ricorrente.
La cheratite erpetica è una delle patologie oftalmiche più severe poiché può determinare un’invalidità permanente del visus per perdita della trasparenza corneale.
La cheratite epiteliale infettiva presenta vescicole corneali che si trasformano in ulcere dendritiche. La sintomatologia è rappresentata da dolore marcato, lacrimazione e fotofobia ma tende a ridursi anche per la comparsa di ipoestesia corneale.
L’ulcera dendritica è una lesione i cui bordi epiteliali contengono il virus. Un allargamento dell’ulcera dendritica determina la cosiddetta ulcera geografica.
Per impostare una terapia adeguata occorre capire quale meccanismo virale è responsabile di una determinata manifestazione oculare. La cheratite erpetica è, infatti, una patologia complessa in cui si intersecano sia meccanismi di replicazione virale attiva sia risposte immunitarie e infiammatorie.
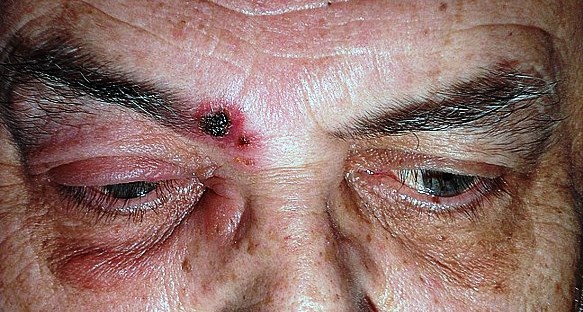
Il virus varicella-zoster (VZV) coinvolge l’occhio in due terzi dei casi, la sua riattivazione, favorita da stress psicofisici e da immunodepressione dà luogo alla malattia secondaria, l’herpes zoster. La riattivazione del VZV a livello della prima branca del trigemino dà luogo all’herpes zoster oftalmico (HZO).
Dopo alcuni giorni compare la tipica eruzione cutanea eritemato-vescicolo-crostosa, di entità molto variabile: solo dolore, poche vescicole, oppure un’ulcerazione massiva con un gran numero di lesioni, anche oculari a congiuntiva, sclera, cornea ed uvea.
Le manifestazioni più ricorrenti sono quelle corneali che possono interessarne tutti gli strati: sono la cheratite puntata superficiale, pseudo dendritica, nummulare, disciforme e provocano arrossamento, dolore, calo visivo e nei casi più gravi alla perdita della trasparenza corneale. L’herpes zoster oftalmico è riconoscibile anche in base all’aspetto caratteristico dell’eruzione cutanea sulla fronte e sulle palpebre.
La terapia antivirale sistemica deve essere prescritta in ogni caso di HZO.
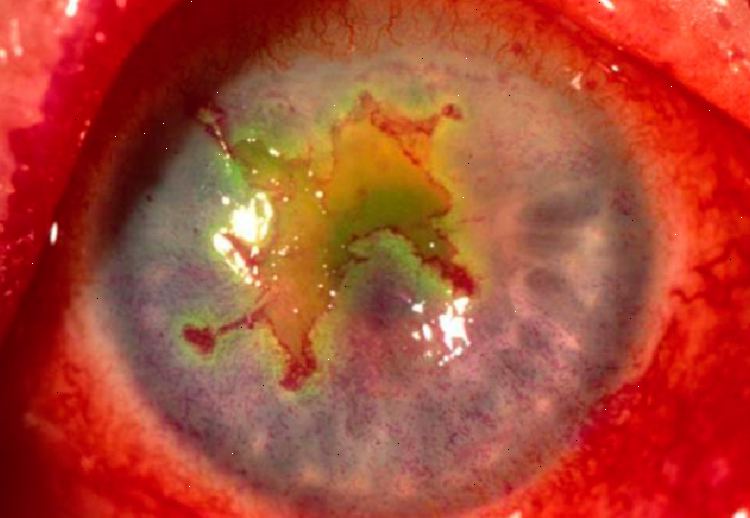
Tipica complicanza dell’ulcera corneale che consiste nell’accumulo di essudato purulento, visibile anche a occhio nudo, che si forma nella camera anteriore a livello della porzione inferiore del limbus. Si presenta come una stria biancastra simile ad una scalfittura.
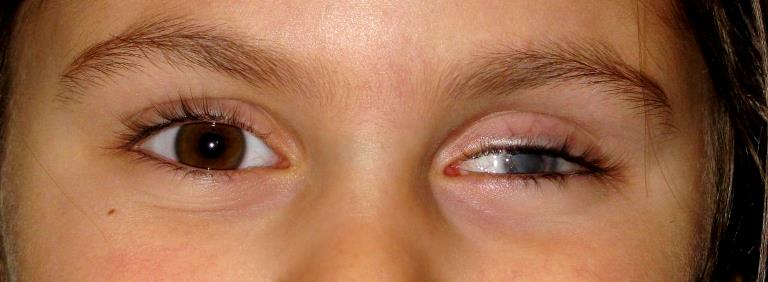
Patologia che si manifesta quando lo sviluppo del nervo ottico e del bulbo si arrestano nelle prime settimane di vita intrauterina.
I termini Ipoplasia e Microftalmo sono spesso sinonimi poiché il microftalmo identifica la presenza alla nascita di un occhio con una lunghezza assiale inferiore a 17 mm (la misura di riferimento alla nascita) anche se la tendenza è quella di definire il bulbo microftalmico solo quando è di volume inferiore ad un terzo della sua dimensione normale.
L’ipoplasia oculare è una patologia rara poiché si presenta in 1,4 casi per 15.000 bambini nati vivi. Il bulbo ipoplasico è quasi sempre parzialmente o totalmente cieco ed è spesso associato ad altri disturbi come lo strabismo ed il nistagmo.
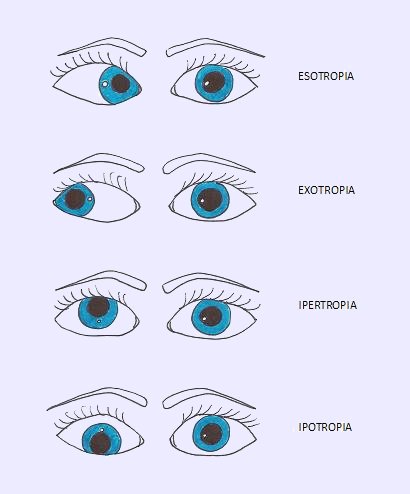
Varietà di strabismo in cui il globo oculare è deviato verso il basso, si contrappone all’ipertropia ed è causata da un’anormalità nel funzionamento del muscolo oculomotore (a carico del muscolo stesso o riguardante il nervo che lo controlla).
Patologia infiammatoria che colpisce sia l’iride che il corpo ciliare, causata nella maggior parte dei casi da una eccessiva risposta immunitaria ad altre patologie o da una reazione allergica. A volte è in associazione a gotta, artrite reumatoide, sarcoidosi, brucellosi ed altre infezioni.
L’iridociclite provoca forte dolore all’occhio oltre a fotofobia, iperemia ed epifora. Nella forma acuta possono formarsi essudati ed aderenze tra iride e cristallino mentre in quella cronica i sintomi sono simili ma di minore entità, anche se con tendenza alla riacutizzazione.
Patologia che consiste nella chiusura incompleta della rima palpebrale per cui il globo oculare rimane parzialmente scoperto. Può essere congenito per una malformazione palpebrale oppure acquisito, a causa della lesione del nervo facciale. L’incompleta chiusura delle palpebre espone la cornea e la congiuntiva alla disidratazione con facile insorgenza di lesioni epiteliali ed ulcere corneali che possono complicarsi in processi infettivi.
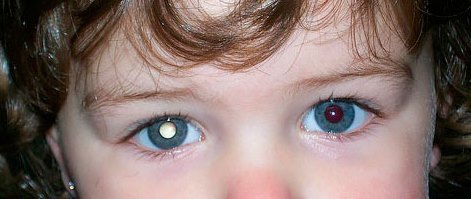
La leucocoria è un riflesso bianco visibile a occhio nudo attraverso la pupilla, più o meno esteso ed intenso ed è il sintomo di una patologia a carico del cristallino, del corpo vitreo o della retina. Le patologie che possono essere associate alla leucocoria sono:
In alcuni rari casi (1%) la leucocoria è causata dal morbo di Coats (fuoriuscita di vasi retinici).
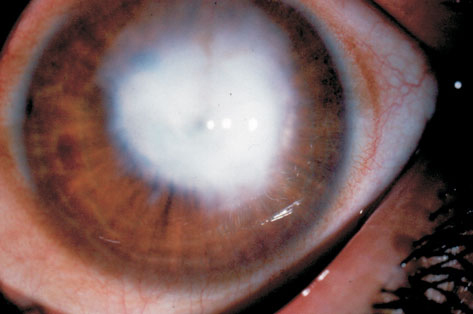
Patologia che consiste nella opacizzazione e perdita della trasparenza corneale dovuta ad un processo di cicatrizzazione in seguito ad un’abrasione corneale profonda (causata da un trauma, da una cheratite o da una patologia infettiva) ed è caratterizzato dalla formazione di cicatrici consistenti in tessuto connettivo biancastro.
Mentre per i piccoli leucomi periferici quasi sempre non è necessaria alcuna terapia, per quelli che interferiscono con la visione occorre intervenire riducendo la lesione chirurgicamente, con interventi tanto più invasivi quanto è maggiore la profondità della lesione stessa.
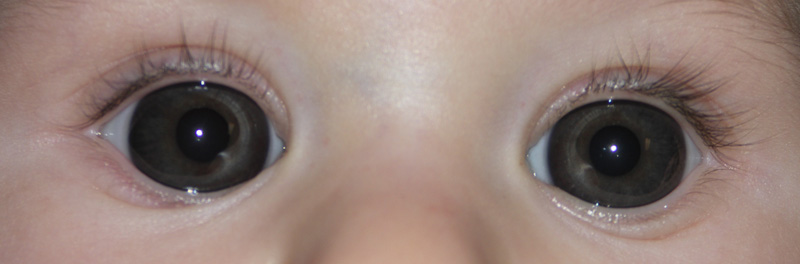
Patologia congenita che consiste nell’aumento delle dimensioni della cornea che oltrepassano i 12 mm. di diametro (anziché i normali 10-11 mm.) e che può essere a sua volta sintomo di glaucoma congenito.
La membrana del Descemet, base su cui poggiano le cellule dell’endotelio corneale, viene spesso lacerata dalla pressione endoculare per cui diventano visibili strie multiple (strie di Haab) orientate in senso orizzontale o curvilineo, concentriche al limbus corneale. L’ipertono oculare induce una miopia di tipo assile e il glaucoma congenito viene denominato in questo caso anche buftalmo, termine che indica la coesistenza di miopia assile e di megalocornea, cioè un incremento complessivo di tutti i diametri oculari.
I neonati con megalocornea a rischio glaucomatoso (rivelano irritabilità e fotofobia) devono essere sottoposti ad esami diagnostici per l’accertamento dell’eventuale danno alla testa del nervo ottico (escavazione papillare) ed alla membrana di Descemet.
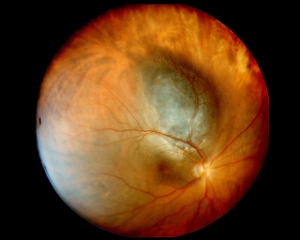
Tumore maligno endoculare primitivo della coroide, più frequente nell’adulto, con un’incidenza pari a 6 casi su un milione, la cui localizzazione è variabile e può interessare il polo posteriore, la zona papillare, la media ed estrema periferia.
L’origine della neoplasia è dovuta a molti fattori e sebbene non siano ancora ben conosciuti i fattori di rischio, è evidente una predisposizione della razza caucasica e un’età compresa tra i 50 e i 60 anni. Quasi sempre asintomatico, il melanoma insorge nella maggioranza dei casi ex novo, mentre in una ridotta percentuale di casi si sviluppa a partire da un neo.
I sintomi sono relativi alla posizione ed al volume del tumore: se al polo posteriore si avrà un rapido calo del visus mentre se in periferia può essere del tutto asintomatico fino a quando non insorgono complicanze (es. distacco di retina).
I piccoli melanomi pigmentati possono essere trattati con la termoterapia transpupillare, metodica di recente introduzione che attraverso un laser a diodi determina un aumento di temperatura dentro il tumore, provocandone la necrosi.
Questa tecnica può essere impiegata anche per melanomi di maggiori dimensioni se associata alla radioterapia con placche episclerali (terapia sandwich). La radioterapia con placche radioattive (brachiterapia), invece, rappresenta attualmente il trattamento radiante più utilizzato.
Il melanoma della coroide però, anche se la terapia conservativa può dare risultati soddisfacenti, rimane una delle più frequenti cause di enucleazione del bulbo oculare.
Questa patologia è inoltre caratterizzata, in molti casi, dallo sviluppo di metastasi a distanza: le sedi preferenziali sono il fegato (92% dei casi), il polmone (31%), lo scheletro (23%), la cute (17%) ed il sistema nervoso centrale (4%). Il tempo di comparsa delle lesioni secondarie è estremamente variabile (da 2 mesi a 30 anni) e spesso la loro comparsa dà una diagnosi infausta.
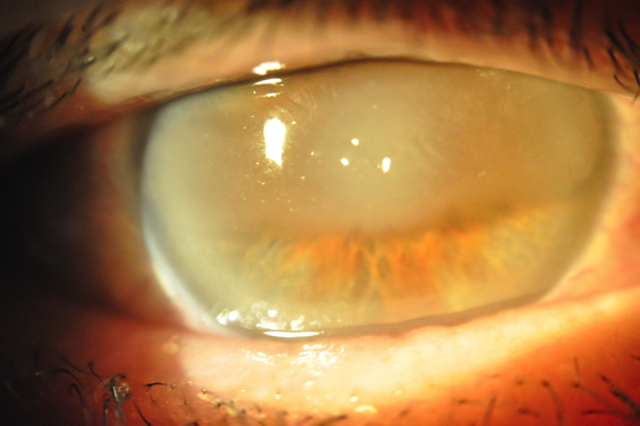
Il melting corneale (o colliquazione corneale) è una grave patologia della cornea che consiste nel suo assottigliamento e nelle forme più gravi nella fusione e liquefazione del tessuto corneale. Se non trattato tempestivamente, può evolvere rapidamente in una perforazione dell’occhio, mettendo a rischio la vista.
Le cause principali che sviluppanol il melting corneale sono:
Le Terapie sono improntate al blocco immediato del processo distruttivo del tessuto corneale e sono di tipo medico con impiego intensivo di colliri antibiotici, antivirali o antinfiammatori mirati oppure gli inibitori delle collagenasi per bloccare gli enzimi che degradano la cornea.
Le Lenti a contatto terapeutiche sono utilizzate successivamente per proteggere l’epitelio e favorire la guarigione mentre l’intervento di Cheratoplastica o l’uso di membrane amniotiche divengono necessari nei casi più gravi o a rischio di perforazione della cornea.
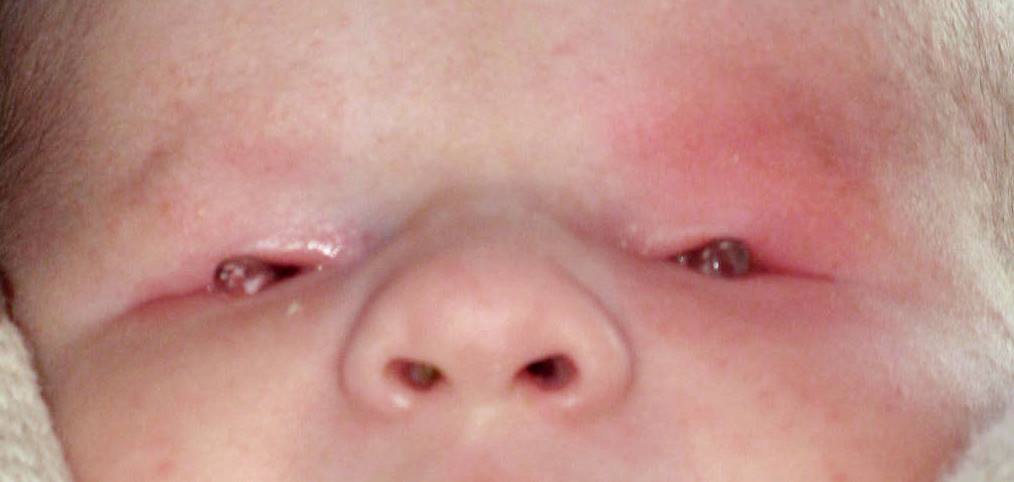
Il microftalmo congenito è una malformazione oculare caratterizzata dallo sviluppo incompleto del bulbo oculare dovuto all’arresto della crescita dell’occhio nelle prime settimane di vita intrauterina. Si presenta con un enoftalmo (occhio infossato) e con la tendenza della palpebra interessata a rimanere più chiusa. Può insorgere sporadicamente oppure manifestare un’ereditarietà dominante ad espressione variabile o recessiva e si presenta in 1,4 casi per 15.000 bambini nati vivi.
Il microftalmo può essere mono o bilaterale ed è rilevabile con la presenza alla nascita di un occhio più piccolo del normale, con lunghezza assiale inferiore a 17 mm, cornea e iride di dimensioni più piccole. In alcuni casi il bulbo è vedente ma più spesso è privo di capacità visiva.
Il microftalmo congenito è spesso associato ad altre patologie congenite quali cataratta congenita, glaucoma, coloboma dell’iride, oppure può associarsi a malformazioni congenite di altri organi. Il microftalmo può essere favorito da fattori di rischio quali: età materna superiore ai 40 anni, indice di massa corporea materno elevato, fumo materno, parto gemellare, neonato di basso peso alla nascita, esposizione prenatale ad agenti teratogeni, alcolismo, oppure per effetto di infezioni contratte dalla madre durante la gestazione (es. toxoplasmosi, herpes simplex, rosolia, citomegalovirus) o ancora a causa di diverse anomalie cromosomiche o malattie genetiche.
I bambini affetti da microftalmo necessitano di un programma di riabilitazione orbitaria protesica per evitare l’instaurarsi di un microrbitismo: l’applicazione della protesi, infatti, stimola l’accrescimento della struttura ossea ma anche dei tessuti molli (palpebre e congiuntiva).
Nei primi mesi di vita viene applicata una protesi su misura che deve essere sostituita ogni 2-3 settimane con altre di dimensioni sempre maggiori allo scopo di aumentare il volume dei tessuti e favorire il corretto funzionamento delle palpebre, mentre nei sei mesi successivi sarà sufficiente sostituire la protesi ogni mese. La corretta applicazione della protesi oculare consente in seguito un risultato estetico soddisfacente poiché il movimento della protesi sarà proporzionale a quello del microftalmo sottostante.
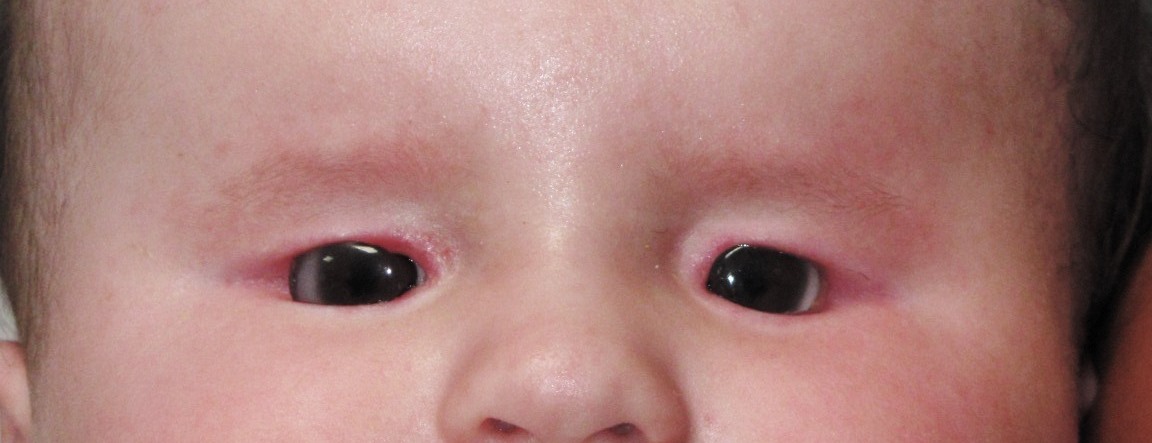
Microrbitismo (orbita piccola), microblefarismo (ipoplasia palpebrale) e ipoplasia emifacciale sono le condizioni tipiche che residuano dall’anoftalmia o microftalmia congenita.
L’orbita di un bambino cresce per il 70% durante i primi tre anni di vita, mentre il restante 30 % si sviluppa nei successivi 5-6 anni. Nei casi più gravi di microftalmia o anoftalmia è necessario perciò instaurare un programma di riabilitazione orbitaria protesica in modo da evitare l’aggravarsi di questo quadro.
L’intervento precoce stimolerà l’accrescimento sia della struttura ossea che dei tessuti molli, palpebre e congiuntiva che sono maggiormente estensibili nelle prime fasi della vita, in armonia con la regione controlaterale, consentendo un risultato estetico più accettabile.

Sono causate da fenomeni degenerativi del corpo vitreo che perde la propria trasparenza per una riduzione della componente acquosa soprattutto in seguito al normale processo fisiologico legato all’invecchiamento ma può essere causato anche dalla miopia.
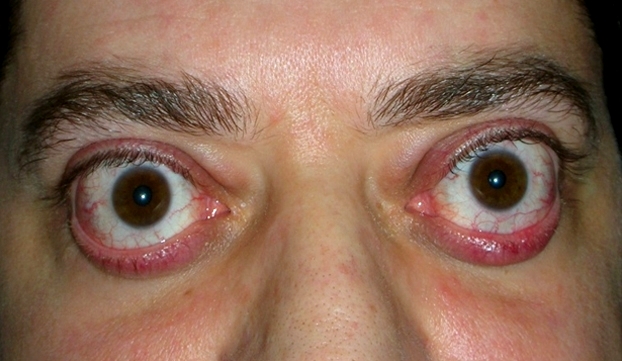
Patologia autoimmune che insorge a qualsiasi età, anche se colpisce soprattutto donne fra i venti e i sessant’anni. Le alterazioni del sistema immunitario sembrano avere un ruolo rilevante nella sua patogenesi.
Nella malattia di Basedow l’organismo produce autoanticorpi che attaccano la ghiandola tiroide, la cute degli arti inferiori e gli occhi. I pazienti affetti dal morbo possono presentare, oltre ai sintomi sistemici caratteristici dell’ipertiroidismo, anche alterazioni cutanee e soprattutto alterazioni oculari.
I sintomi più evidenti sono: gozzo pronunciato, esoftalmo con sguardo fisso, scarso ammiccamento, retrazione palpebrale, chemosi e rossore congiuntivale che producono dolore, lacrimazione abbondante, irritazione delle mucose e spesso anche fotofobia.
Il bulbo oculare tende a protrudere all’esterno dell’orbita a causa dell’accumulo di liquido e di grasso nella zona retrobulbare. Gli autoanticorpi infatti, infiltrano i tessuti dei muscoli estrinseci ed il grasso orbitario, causando deficit di mobilità e retrazione palpebrale che possono degenerare in una cheratite da esposizione.
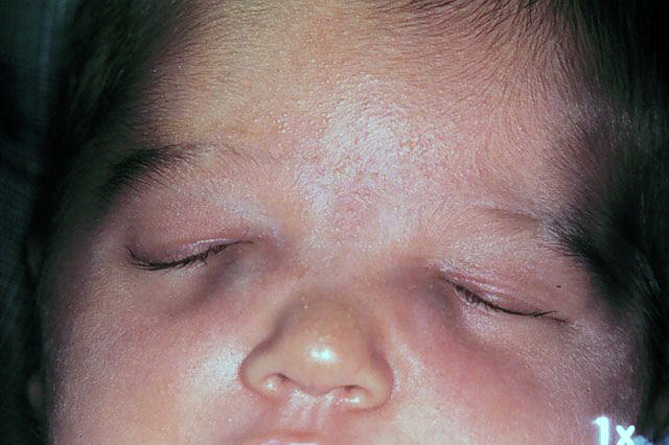
Formazione cistica proveniente dai seni paranasali ed è secondaria ad accumulo di muco causato dalla ostruzione delle aperture di queste cavità che in pratica sequestrano il materiale mucoso (cisti da ritenzione). La massa cistica ripiena di muco prodotto all’interno della lesione è delimitata da uno strato osseo. Frequentemente si espande nell’orbita per erosione della parete ossea. L’occlusione può essere dovuta ad un processo infiammatorio, può avere origine post-traumatica o essere causata da anomalie anatomiche o tumori. L’incidenza è maggiore negli adulti fra i 50 e i 70 anni ed è localizzata nella maggior parte dei casi nel seno frontale e nel seno etmoidale mentre i seni mascellari e i seni sfenoidali sono interessati raramente.
I sintomi più frequenti sono l’esoftalmo, la protrusione o dislocazione del bulbo, sensazione di dolore orbitale, diplopia e in qualche caso danni al nervo ottico. Il mucocele normalmente ha una crescita lenta e può svilupparsi dopo molti anni anche senza presentare sintomi. In alcuni casi può interessare anche le vie lacrimali e presentarsi come una tumefazione dell’area del sacco lacrimale e della radice del naso.
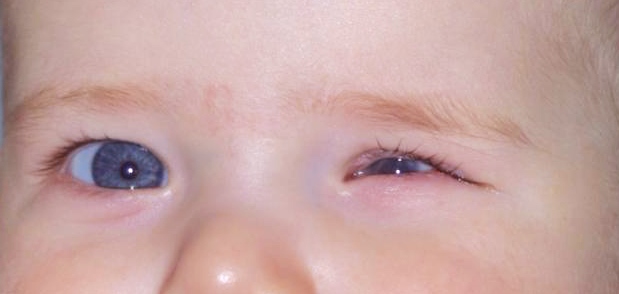
E’ così definito il microftalmo congenito quando non esistono altre patologie o sindromi correlate e concomitanti. In genere è presente glaucoma ad angolo chiuso. Il nanoftalmo è geneticamente eterogeneo: ne esistono due forme autosomiche dominanti (NNO1 e NNO3) e una forma autosomica recessiva (NNO2), causata da mutazioni nel gene MFRP.
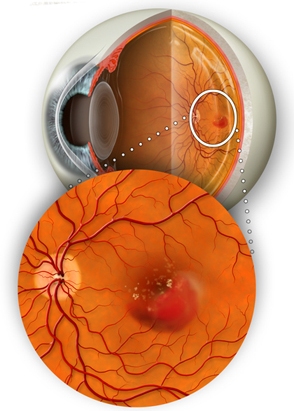
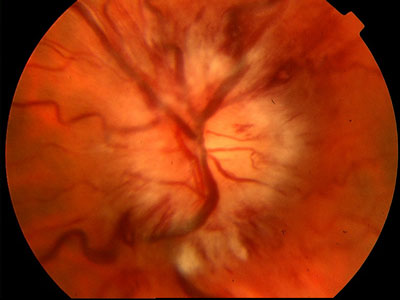
Molte patologie oculari hanno patogenesi neovascolare, derivano cioè dalla crescita anomala di capillari all’interno dell’occhio e spesso rappresentano una importante causa di cecità o ipovisione. Lo sviluppo di neovasi patologici nella maggior parte dei casi è dovuto all’ipossia ed alla scarsa perfusione retinica.
Nella forma “umida” della degenerazione maculare senile (AMD), i neovasi si sviluppano dietro la retina, causando il sollevamento della macula dalla sua posizione normale e determinando visione confusa e distorta. Il danno maculare si sviluppa rapidamente e in mancanza di terapia porta rapidamente alla perdita della visione.
La crescita anomala di neovasi all’interno dell’occhio è riscontrabile nella miopia elevata, nella retinopatia diabetica proliferante, nelle occlusioni venose, nel glaucoma neovascolare, nei traumi oculari. Il punto comune tra queste patologie sembra essere l’ischemia a livello retinico ma a volte la neovascolarizzazione avviene senza cause evidenti e riconoscibili.
Neovasi vengono generati anche nell’edema maculare refrattario, causato dal rigonfiamento dell’area maculare per accumulo di liquido sottoretinico, una forma di edema che non risponde adeguatamente alle terapie normalmente in uso, riducendo la visione. Può verificarsi in condizioni quali l’occlusione della vena centrale retinica, la retinopatia diabetica, dopo impianto di lente intraoculare (IOL). In assenza di terapia efficace, la perdita di visione può progredire e divenire permanente.
La formazione di nuovi vasi è un processo biologico fondamentale: è necessaria all’embriogenesi, allo sviluppo della placenta ed alla crescita normale dei tessuti e degli organi, nonché alla guarigione delle ferite. Per contro, è alla base della crescita tumorale e di altre patologie. La formazione di neovasi (angiogenesi) sembra avvenire nello stesso modo sia in caso di sviluppo normale che patologico. I vasi neoformati presentano caratteristiche anomale e spesso dannose che sono alla base delle manifestazioni cliniche delle malattie neovascolari dell’occhio:
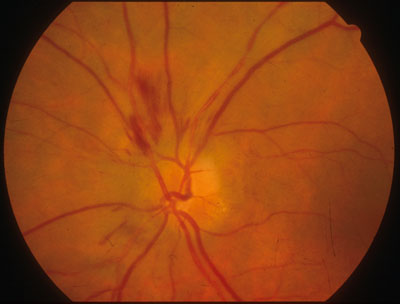
Patologia infiammatoria a carico della parte retro-bulbare del nervo ottico, spesso bilaterale e di origine autoimmune, consiste nella reazione immunitaria contro la guaina mielinica che riveste il nervo ottico e che impedisce al segnale elettrico da esso trasmesso di deteriorarsi.
Può essere il sintomo d’esordio di una malattia demielinizzante come la sclerosi multipla o a placche che in questi casi, oltre al nervo ottico, colpisce la guaina mielinica nel suo complesso, compreso il cervello. I sintomi tipici sono la a rapida perdita della vista e il dolore nel muovere gli occhi.
La remissione spontanea, con il ripristino del visus normale si può verificare in uno-due mesi ma in alcuni casi rimane uno scotoma centrale e nella maggior parte dei casi non si arriva ad un ritorno alla normalità.
Sono frequenti infatti le recidive, specialmente nella sclerosi multipla. Ogni ricaduta aumenta il danno alla vista residua: le conseguenze possono essere atrofia ottica e la cecità permanente.
Noduli di Lisch
Neurofibroma plessiforme
Patologia genetica del gruppo delle facomatosi caratterizzato dallo sviluppo di tumori benigni sui nervi, può colpire anche la pelle e provocare deformità ossee. Esistono due forme: neurofibromatosi di tipo 1 (NF1) e neurofibromatosi di tipo 2 (NF2). La schwannomatosi è considerata una variante della NF2. La forma più comune è la NF1, che provoca tumori benigni detti neurofibromi nei tessuti e negli organi. È una delle malattie genetiche più diffuse: colpisce 1 persona su 3500.
Poiché, in alcuni casi, i neurofibromi possono diventare maligni, la gestione della malattia è incentrata sul controllo delle eventuali alterazioni di questi tumori. A livello sistemico può generare neoplasie a livello dei nervi periferici e del sistema nervoso centrale, anomalie scheletriche, fibromi tipo mollusco e può essere associata ad ipertensione e ritardo mentale.
A livello oculare si possono associare noduli iridei di Lisch, neurofibromi palpebrali, glioma del nervo ottico ed ectropion congenito dell’uvea che può a sua volta essere associato a glaucoma. Più raramente si possono sviluppare astrocitomi retinici e meningiomi.
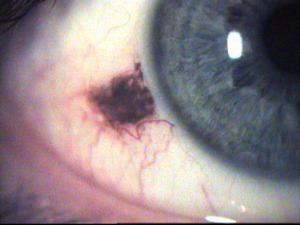
Piccola lesione pigmentata di origine benigna localizzata in genere sulla congiuntiva bulbare (nevo congiuntivale) che può formarsi anche all’interno del bulbo, sulla coroide (nevo coroideale) e in rari casi (uno su 5.000) può trasformarsi in un melanoma maligno.
I nevi devono essere controllati almeno ogni anno e se non presentano variazioni di forma, volume o colore e se non intefreriscono con le funzioni del bulbo non devono essere rimossi ed hanno di norma un decorso benigno.
Oscillazione ritmica e involontaria dei bulbi oculari in cui ogni ciclo di movimento tende ad allontanare l’oggetto che si vuole osservare dalla fovea (centro retinico), facendolo uscire dalla zona centrale del campo visivo. Si presenta con un movimento pendolare, con la stessa velocità in tutte le direzioni, o con un movimento irregolare con velocità diversa, composto di una fase rapida e di una fase lenta.
Il nistagmo spesso riduce la visione in modo importante, tanto che molti pazienti sono considerati ipovedenti poiché la profondità di campo diminuisce, provocando instabilità nei movimenti. Tuttavia, la capacità visiva può variare durante il giorno ed essere influenzata da fattori emozionali e fisici come lo stress o la stanchezza.
Il nistagmo patologico può essere dovuto a cause genetiche o acquisito in seguito ad una patologia. Si può manifestare per una lesione del sistema nervoso (cervelletto e tronco encefalico) o dell’apparato vestibolare (organo dell’equilibrio).
Il nistagmo congenito si manifesta in genere entro i primi 2-3 mesi dalla nascita e le cause possono essere varie, cataratta congenita, strabismo, ecc.). Il nistagmo in alcuni casi può presentarsi senza una causa nota e può insorgere anche in persone perfettamente sane, soprattutto in caso di stress e affaticamento.
.
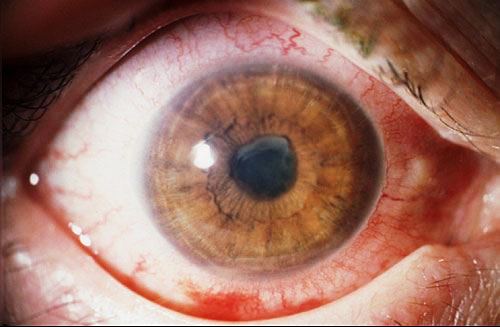
E’ una complicanza rara e devastante che consiste nell’insorgenza di una uveite riflessa nell’occhio sano e compare nei mesi successivi ad un trauma perforante con prolasso di tessuto uveale o ad un intervento chirurgico al bulbo, soprattutto nel polo posteriore. La patologia conduce in breve tempo alla formazione di una panuveite granulomatosa.
L’occhio traumatizzato è detto “eccitante” mentre l’altro è detto “simpatizzante“, i sintomi sono: dolore, fotofobia, paresi dell’accomodazione, metamorfopsia e perdita del visus, da moderata a significativa. L’infiammazione può causare distacco retinico e edema maculare grave. I sintomi extraoculari possono comprendere cefalea, meningite e perdita dell’udito.
L’eziologia dell’OS non è ancora chiara, si pensa sia dovuta ad un meccanismo immunitario e alla risposta infiammatoria autoimmune diretta contro gli auto-antigeni oculari che si formano a seguito della lesione iniziale.
Il trattamento chirurgico resta controverso e l’enucleazione o l’eviscerazione devono essere proposte solo nel caso di cecità all’occhio colpito, dato che la prognosi visiva dell’occhio controlaterale è variabile. È stato osservato che l’enucleazione tardiva non porta benefici.
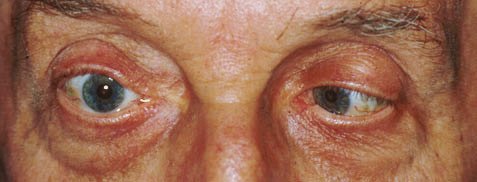
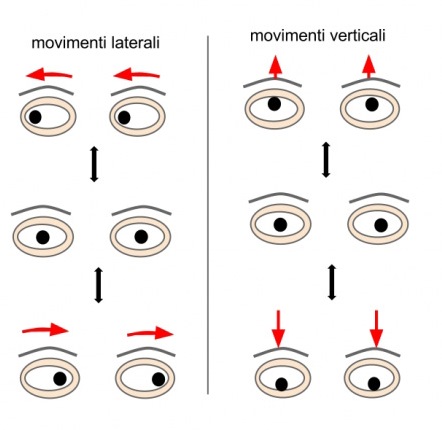
Paralisi della muscolatura estrinseca o intrinseca del bulbo che impedisce i normali movimenti del bulbo stesso, compresi quelli involontari dell’accomodazione. Le cause sono molteplici: traumi, affezioni di origine infiammatoria o infettiva, lesioni vascolari o tumori.
L’oftalmoplegia internucleare bilaterale in soggetti giovani è quasi sempre associata a sclerosi multipla mentre quella monolaterale è in genere secondaria a lesioni vascolari o ischemiche.
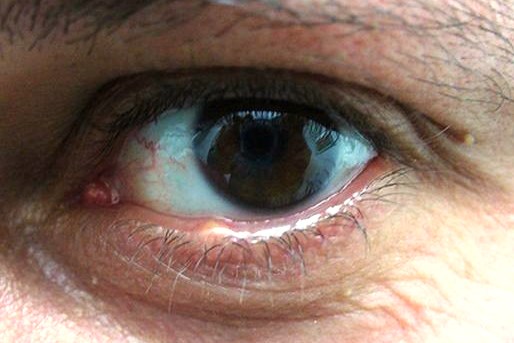
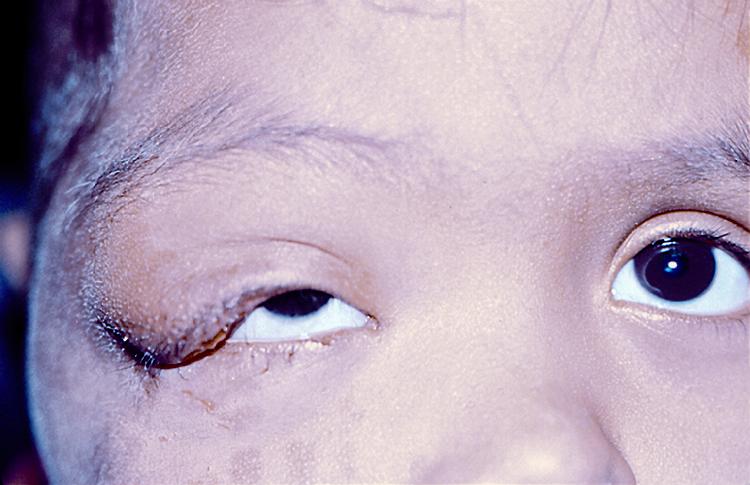
L’orzaiolo è un’infiammazione acuta che colpisce le ghiandole sebacee situate sul bordo delle palpebre e può formarsi all’esterno della palpebra quando è interessata una ghiandola di Zeis o di Moll oppure all’interno, se colpisce una ghiandola di Meibomio. La causa più frequente è un’infezione batterica da staf
La guarigione è solitamente spontanea ma nei casi più dolorosi o in presenza di una cronicizzazione può essere effettuata una terapia con pomata antibiotica.
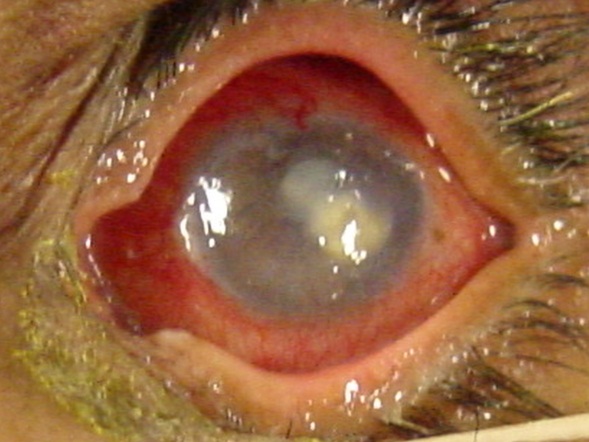
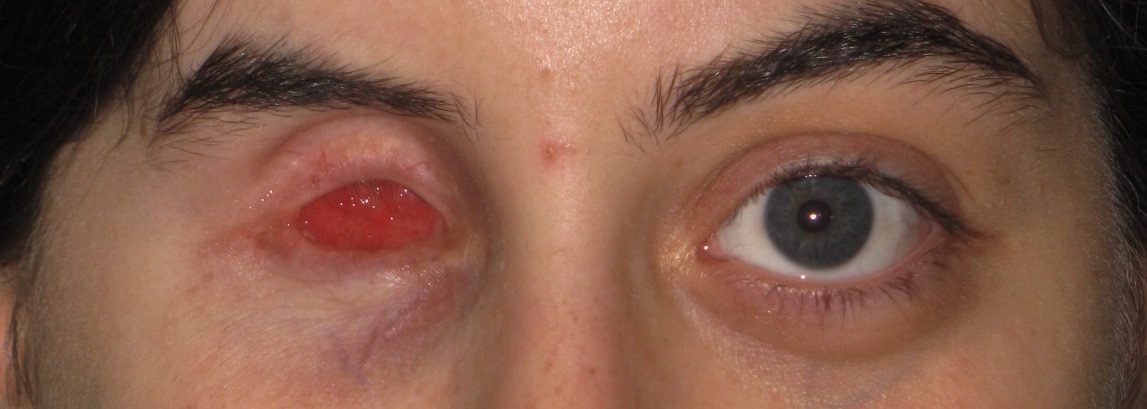
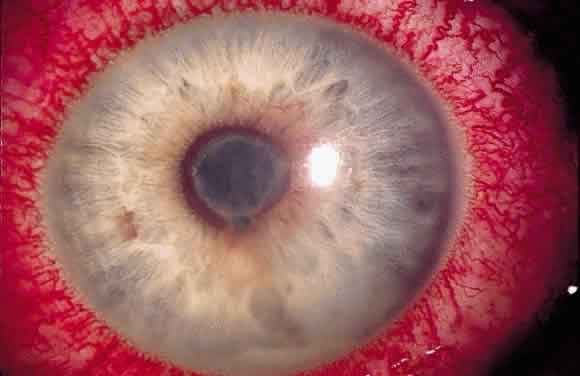
Vasta infezione endobulbare dal quadro clinico drammatico caratterizzata da una primitiva infiammazione purulenta dell’uvea e successivamente dell’intero bulbo oculare, provocata o da lesioni esterne dell’occhio (forma diretta) o da emboli settici (forma metastatica). Se non è curata tempestivamente, provoca la colliquazione purulenta delle parti interne del bulbo. E’ caratterizzata da dolore intenso, edema congiuntivale e palpebrale, esoftalmo ed essudato purulento in camera anteriore.
La pupilla, se visibile, lascia intravedere infiltrati purulenti anche nel vitreo. Il quadro evolve rapidamente in una perforazione del bulbo e ftisi.
Le cause possono essere infezioni esterne (batteriche o fungine per esempio in caso di traumi perforanti, corpi estranei endobulbari, interventi chirurgici) o da strutture vicine (processi suppurativi orbitari o dei seni paranasali) oppure dall’interno dell’organismo per via ematica (endocardite settica, meningococcemia, sepsi). La patologia nella sua fase terminale provoca la perdita della vista e richiede l’enucleazione del bulbo.
Flogosi che interessa l’intera membrana uveale, sia anteriore che posteriore. Le forme acute sono rappresentate dalle endoftalmiti, secondarie a diffusione metastatica di infezioni batteriche, micotiche e virali. Si possono però osservare anche delle endoftalmiti acute sterili, espressione di reazioni di ipersensibilità a materiali endogeni o esogeni.
Le panuveiti croniche sono invece costituite dalle irido-ciclo-coroiditi o uveiti diffuse e ricorrono nell’evoluzione di uveiti anteriori o posteriori gravi protratte o in corso di malattie generali, batteriche e parassitarie.
Le principali forme di panuveite sono:
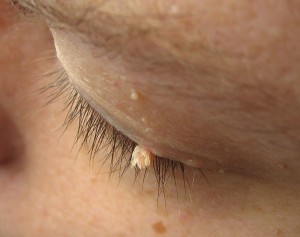
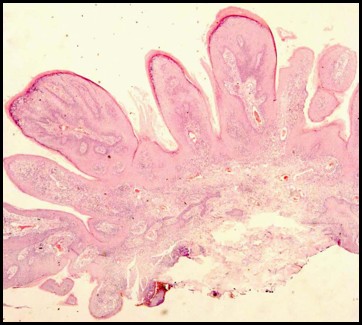
Lesione epiteliale benigna della cute o della mucosa formata da piccole papille che può coinvolgere le palpebre, la congiuntiva e la regione periorbitale. E’ localizzato a livello del fornice congiuntivale (papilloma congiuntivale) oppure sul margine libero della palpebra (papilloma palpebrale), può essere di forma piatta o peduncolata ed ha spesso cause virali, quindi trasmesse per contagio diretto.
La rimozione delle forme cutanee, sia essa effettuata con modalità tradizionale (bisturi e cauterizzazione), criogenica (con azoto liquido o applicazioni dirette di freddo) o laser non è mai dolorosa, a patto che si ricorra a tecniche anestetiche appropriate.
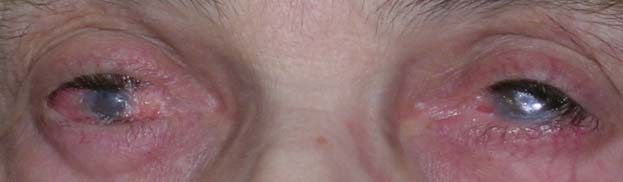
Patolgia rara che consiste nella localizzazione oculare di un gruppo di malattie autoimmuni sistemiche di tipo bolloso che hanno come obiettivo la membrana basale della pelle e delle mucose (congiuntiva, cavo orale, naso, esofago, laringe, genitali, vie urinarie). Colpisce uomini e donne in una proporzione di uno a tre in una età compresa tra i 30 e i 90 anni, con un picco massimo verso i 70 anni.
Quando la bolla scoppia crea una cicatrice che ha come effetto secondario la retrazione del tessuto. Lo stadio terminale del processo infiammatorio autoimmune del pemfigoide oculare è rappresentato dalla cicatrizzazione completa della congiuntiva e dalla cheratinizzazione e neovascolarizzazione della cornea. Questa patologia è curabile se trattata tempestivamente altrimenti provoca cecità bilaterale irreversibile.
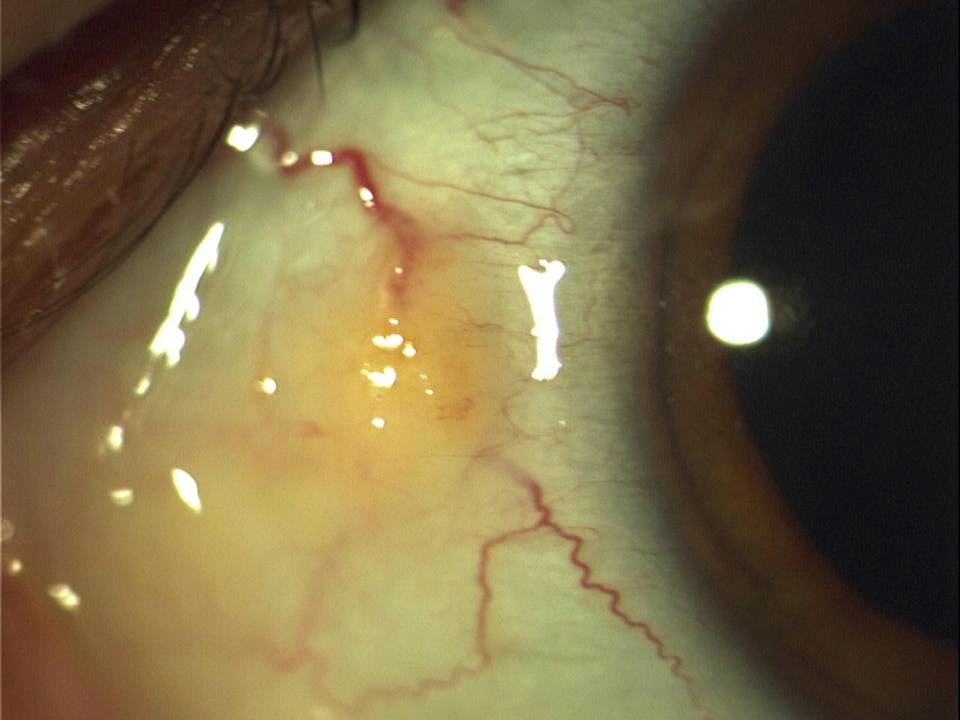
E’ una lesione che si forma sul tessuto superficiale della sclera vicino all’angolo corneale. Di colore giallognolo e leggermente sporgente, è molto comune tra le persone di mezza età o più anziane che passano molto tempo al sole ma si può trovare anche in persone più giovani e persino nei bambini.
Normalmente causa pochi sintomi ma una pinguecola irritata può creare una sensazione di un corpo estraneo e in alcuni casi può infiammarsi ed edemizzarsi. Nei casi gravi la rimozione chirurgica della pinguecola e consigliata soprattutto se crea problemi alla vista, all’uso di lenti a contatto, o perfino alla chiusura delle palpebre. Di frequente, la pinguecola porta alla formazione di pterigio.
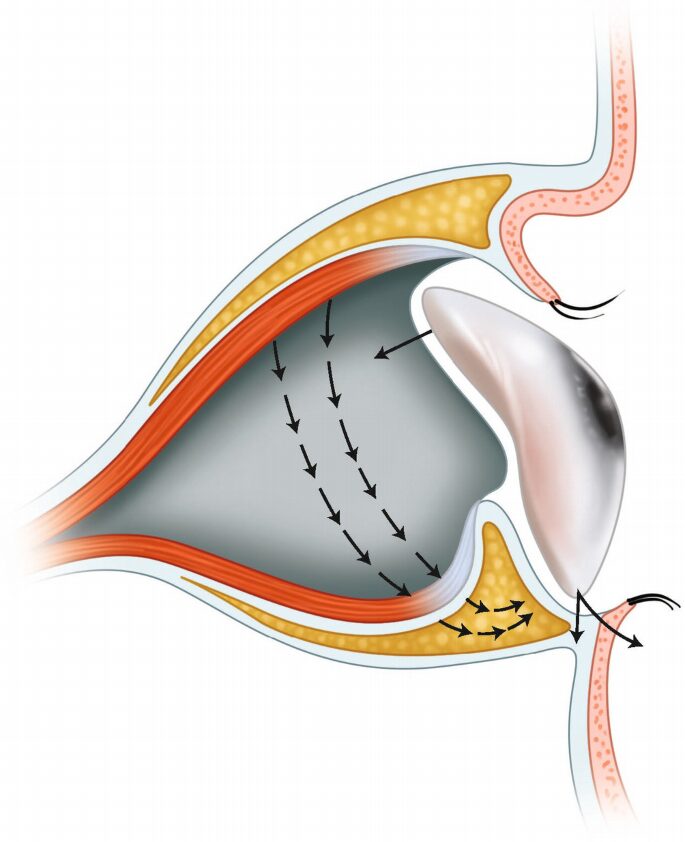
La PESS è una sindrome multifattoriale e variabile causata da uno spostamento rotatorio del contenuto orbitario, unitamente alla retrazione dei muscoli extraoculari e al possibile riassorbimento dell’impianto orbitario, qualora questo sia realizzato in idrossiapatite.
Il meccanismo fisiopatologico principalmente ipotizzato per la PESS era l’atrofia dei tessuti orbitari, in particolare del tessuto adiposo, con conseguenti manifestazioni cliniche variabili. Studi più recenti, condotti con tomografia computerizzata ad alta risoluzione e risonanza magnetica o mediante analisi istopatologiche, non hanno riscontrato atrofia del tessuto adiposo orbitario, bensì uno spostamentomovimento rotatorio dei tessuti orbitari da superiore a posteriore e da posteriore a inferiore, unitamente alla retrazione dei muscoli extraoculari e a una possibile perdita di volume dell’impianto orbitario per riassorbimento, se questo è realizzato in idrossiapatite.
La PESS determina un’inclinazione posteriore del fornice superiore, un solco superiore profondo (enoftalmo), la pseudoptosi della palpebra superiore, un allungamento e una lassità di quella inferiore e la scarsa profondità del fornice inferiore.

Protrusione anomala del bulbo oculare causata da alterazioni localizzate nello spazio retrobulbare o a volte da un’orbita poco profonda. Può essere di origine traumatica o secondaria a processi infiammatori cronici, malformazioni vascolari o tumori a localizzazione orbitaria o endobulbare e successivamente invadenti l’orbita.
Nella sua forma assiale è determinata da lesioni invadenti e localizzate all’interno del cono muscolare, come l’emangioma cavernoso e le neoplasie del nervo ottico o l’oftalmopatia tiroidea.
Lesioni situate all’esterno del cono muscolare danno luogo a una proptosi eccentrica la cui direzione è determinata dalla sede della lesione: può essere monolaterale o bilaterale, simmetrica o asimmetrica.
Nella sua forma acuta, se accompagnata da dolore e chemosi congiuntivale è di solito sintomo di una cellulite orbitaria o pseudotumor infiammatorio. Viene misurata con l’exoftalmomtro e una differenza di 2 mm fra i due occhi è già indicativa e fa propendere per una diagnosi di proptosi. La proptosi viene classificata in lieve (21÷23 mm), moderata (24÷27 mm) e grave (28 mm o più).
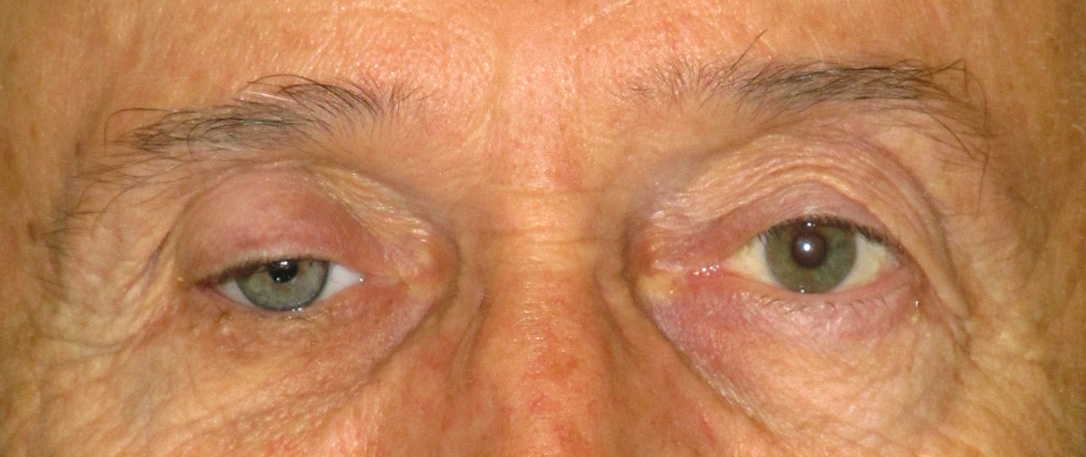
Ptosi “apparente” che può risultare da un deficit volumetrico come quello associato al microftalmo o alla sindrome della cavità anoftalmica. Può essere rilevata anche in diverse altre condizioni come l’eccesso di cute sulla palpebra superiore o l’ipotropia.
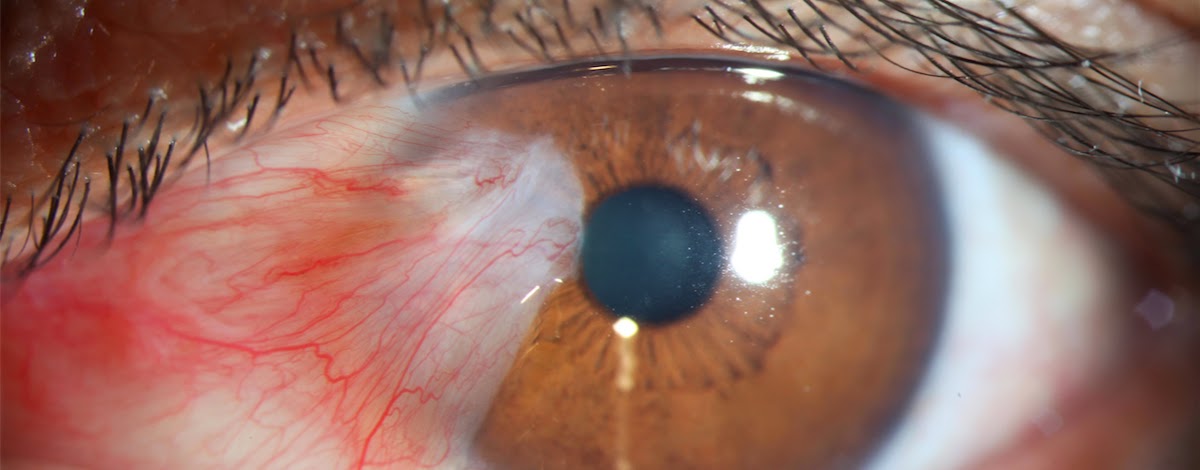
E’ un’escrescenza cuneiforme fibrovascolare che si forma tipicamente sul tessuto superficiale della sclera ma in alcuni casi anche sulla cornea e interferire con la vista. La causa della sua formazione è l’esposizione prolungata ai raggi ultravioletti del sole.
Nella fase iniziale non si avverte alcun sintomo e non è necessaria alcuna terapia ma alcune forme possono causare edema e forte infiammazione, mentre altre possono crescere o assottigliarsi. In questi casi si può avvertire la sensazione di corpo estraneo.
Nella sua forma avanzata può causare una distorsione della superficie corneale e generare astigmatismo. Se la lesione è infiammata e di piccole dimensioni un gel lubrificante può dare sollievo ma in alcuni casi e’ necessaria la sua rimozione chirurgica, tenendo conto che questa lesione è recidivante nel 30-40 per cento dei casi.
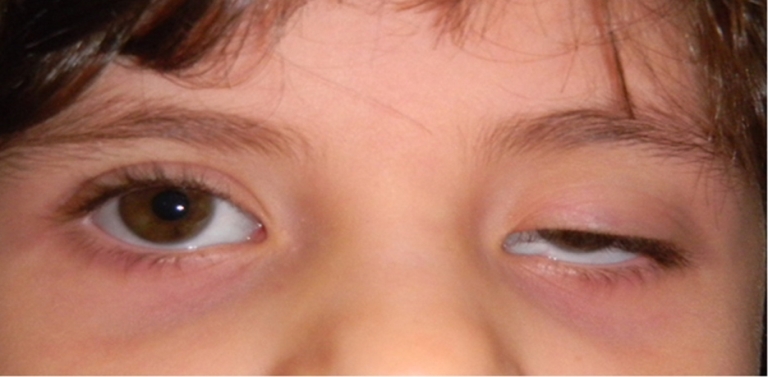
Patologia che consiste nella chiusura della palpebra superiore per l’impossibilità di sollevarla in seguito ad insufficienza funzionale del muscolo elevatore palpebrale. La forma più comune è quella congenita, una distrofia od aponeurosi isolata mono o bilaterale. Esistono anche ptosi acquisite che possono essere:
Se la ptosi è contenuta pone solo problemi di tipo estetico ma se è molto accentuata può interferire con la visione, richiedendo la terapia chirurgica. Nelle ptosi congenite di una certa entità occorre comunque intervenire velocemente poiché possono impedire il corretto sviluppo della funzione visiva.
Raro tumore maligno pediatrico delle parti molli in età pediatrica che rappresenta circa il 3-4% di tutti i tumori infantili. Il nome è formato dalla combinazione di tre parole più piccole: “rabdo”, che significa a forma di bacchetta, “mio” che sta per muscolo, e “sarcoma”. Infatti le cellule del rabdomiosarcoma hanno caratteristiche che ricordano le cellule muscolari dell’embrione.
Colpisce i bambini da 1 a 5 anni ma anche gli adolescenti e più raramente ne possono essere affetti anche gli adulti. Può nascere in qualsiasi parte del corpo, ma generalmente è più frequente a livello di testa-collo (40%), apparato genitourinario (20%) , arti e tronco (20%).
La causa di questo tumore non è nota ma un certo numero di bambini con rabdomiosarcoma ha anche malformazioni congenite di altri organi (cuore, intestino, cervello, etc.) o rare malattie genetiche, come la neurofibromatosi di tipo I.
In generale si rivela come una massa in crescita più o meno rapida, spesso compare una tumefazione o di un nodulo. I tumori che insorgono nella regione della testa e/o del collo possono causare mal di testa, nausea, vomito, diplopia, paralisi facciale e ostruzione delle vie aeree.
E’ distinto in due sottotipi, quello embrionale e quello alveolare.
Il rabdomiosarcoma embrionale è il più frequente e tende a svilupparsi a livello della testa e del collo, della vescica, della vagina nelle femmine e attorno alla prostata e ai testicoli nel maschio.
In pesenza di un rabdomiosarcoma localizzato (cioè senza metastasi) le possibilità di guarigione arrivano fino al 70% ma in circa il 20-25% dei casi Il rabdomiosarcoma si presenta con metastasi già al momento della diagnosi. Gli organi più frequentemente colpiti da metastasi sono i polmoni, le ossa, il midollo osseo, i tessuti sottocutanei.

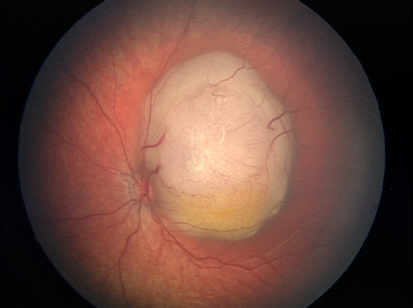
Il retinoblastoma è un tumore maligno della retina tipico dell’età pediatrica e rappresenta il 4% dei tumori pediatrici. Ogni anno colpisce un bambino ogni ventimila nati vivi e può essere monolaterale o bilaterale e frequentemente ha origine ereditaria. I primi sintomi del retinoblastoma vengono rilevati entro il terzo anno d’età e sebbene si tratti di un tumore raro è una delle patologie maligne più diffuse dell’infanzia.
Il retinoblastoma è causato da una mutazione genetica che causa la proliferazione incontrollata di cellule e lo sviluppo del tumore all’interno dell’occhio. L’ereditarietà è principalmente autosomica dominante: circa il 25% dei pazienti presenta una forma bilaterale, che è sempre ereditaria, il 15% dei pazienti è affetto da forma ereditaria monolaterale e il 60% da una forma monolaterale non ereditaria.
I sintomi tipici che devono far sospettare l’insorgenza del retinoblastoma sono la leucocoria (un riflesso bianco nella pupilla dovuto alla massa tumorale retinica che occupa il corpo vitreo), correlata spesso allo strabismo derivato dalla deviazione degli assi oculari, dovuta alla perdita di fissazione dell’occhio patologico.
Il trattamento del retinoblastoma prevede l’approccio terapeutico che però dipende dalle dimensioni del tumore (se meno di 3 mm di dimensione), dalla diffusione alle aree circostanti e dalla funzionalità dell’occhio. Nei casi meno gravi il trattamento può comprendere la fotocoagulazione, la crioterapia e la radioterapia. Nei casi in cui è possibile conservazione della funzione visiva può essere praticata la terapia conservativa per il salvataggio dell’occhio con l’utilizzo di farmaci chemioterapici e trattamenti di controllo locale.
Viceversa, il retinoblastoma di grandi dimensioni con estensione della neoplasia può venire asportato solo mediante enucleazione del globo oculare con contestuale rimozione della maggior parte possibile del nervo ottico. La chemioterapia viene utilizzata a volte per ridurre il volume della massa tumorale o per trattare neoplasie che si sono diffuse oltre il bulbo ma in questi casi più gravi la sola chemioterapia può raramente consentire la guarigione del paziente.
La prognosi del retinoblastoma è complessa: se il tumore viene trattato quando è ancora confinato all’interno del bulbo, oltre il 90% dei pazienti può guarire. La prognosi per i pazienti con lesioni metastatiche è invece decisamente infausta ed il 70% dei pazienti che sviluppano un secondo tumore possono esserne colpiti anche fino a trent’anni dopo la lesione originaria.
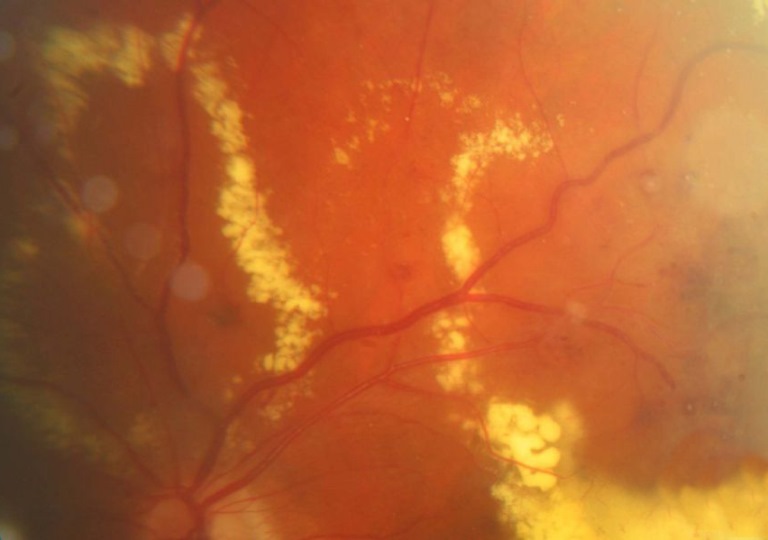
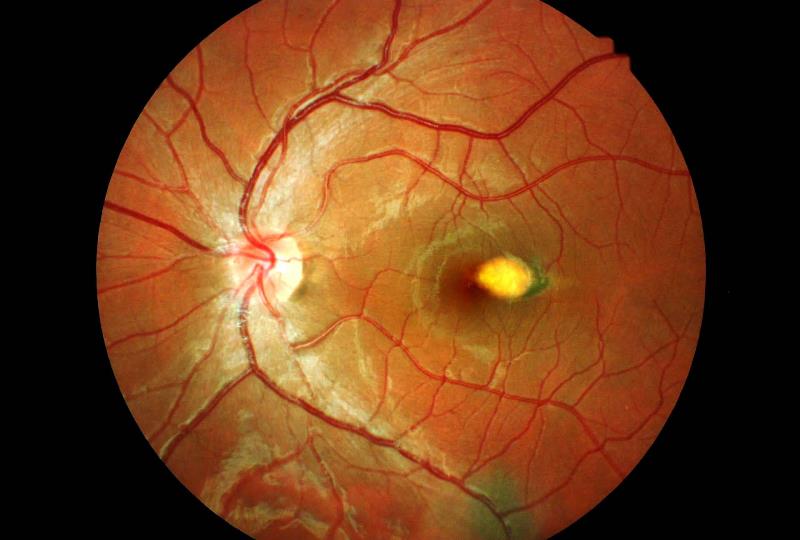
Retinite massiva caratterizzata da un anomalo sviluppo dei vasi sanguigni della retina (telangectasia), con progressiva deposizione di essudati intraretinici e sottoretinici che portano potenzialmente al distacco essudativo della retina.
La patologia è spesso monolaterale e può complicarsi con l’insorgere di un distacco di retina, un glaucoma secondario o una cataratta complicata. Si presenta prevalentemente in bambini di sesso maschile verso la fine della prima decade di vita, non è ereditaria ed il suo decorso clinico è variabile ma quasi sempre progressivo.
La retinopatia di Coats può associarsi ad altre malformazioni vascolari come angiomi facciali, sindrome di Alport, retinopatia pigmentosa ed altre.
Stadiazione:
Fase 1, caratterizzata solo da teleangectasie
Fase 2, presenta teleangectasie ed essudazione con coinvolgimento della fovea
Fase 3, totale o parziale distacco della retina
Fase 4, totale distacco della retina con glaucoma
Fase 5, cecità, occhio indolore o doloroso, spesso con cataratta ed eventuale tisi tubulare
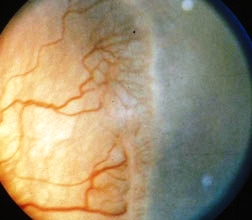
Patologia della retina spesso bilaterale che si manifesta nei nati prematuri con insorgenza intorno alle 32-34 settimane di gestazione con rischio maggiore quanto minore è il tempo di gestazione e quanto è più basso il peso alla nascita. La vascolarizzazione retinica fetale origina dalla papilla e finché questa non è completa rimane sensibile alle elevate concentrazioni di ossigeno tipiche del trattamento in incubatrice.
La retinopatia del prematuro si verifica quando i vasi retinici si sviluppano in modo anormale, formando una cresta di tessuto tra retina centrale vascolarizzata e retina periferica, non vascolarizzata. Nella retinopatia del prematuro grave, questa neovascolarizzazione invade il vitreo.
La patologia provoca la formazione di neovasi nella periferia retinica e successivamente emorragia vitreale, fibrosi e trazione retinica che nello stadio più avanzato si trasforma in fibroplasia retrolentale. La sua evoluzione può portare alla cecità in seguito a distacco per trazione.
I bambini con una retinopatia del prematuro risolta hanno un rischio più alto di miopia, di strabismo e soprattutto di ambliopia, nonché di lesioni cicatriziali e sono a rischio di distacco di retina o, più raramente, di glaucoma e cataratta.
Allo stadio iniziale la terapia consiste nel criotrattamento o nel trattamento laser, “bruciando” la retina periferica per evitare che possa fornire degli stimoli per la formazione dei nuovi vasi dannosi. Quando si manifestano gli ultimi stadi della malattia è necessario intervenire chirurgicamente con vitrectomia o piombaggio sclerale. La R.O.P. è una delle maggiori cause di ipoplasia del bulbo nei bambini.
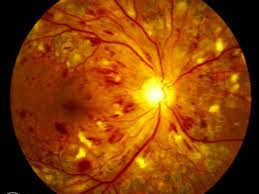
Questa patologia, importante causa di cecità, può essere particolarmente grave nei pazienti affetti da diabete mellito insulino-dipendente (DID) di tipo 1. Si manifesta spesso anche nel diabete mellito cronico non insulino-dipendente (DNID) di tipo 2. Il grado della retinopatia sembra strettamente correlato con la durata del diabete.
Il diabete colpisce il microcircolo retinico anche se nelle prime fasi della malattia non si evidenziano grosse alterazioni dei vasi. I primi segni della retinopatia diabetica sono spesso rappresentati dalle dilatazioni venose e da piccole macchie rosse (microaneurismi capillari) che si osservano all’esame oftalmoscopico nel polo retinico posteriore.
Le emorragie retiniche, l’edema profondo e gli essudati lipidici possono alterare la funzione maculare. I sintomi tardivi sono una diminuzione generalizzata del visus provocata da edema maculare.
L’edema maculare è una causa comune di alterazione visiva nei diabetici e si manifesta con la comparsa di essudati molli (microinfarti causati da ridotta perfusione retinica) ed essudati duri che invece sono provocati da un edema cronico.
La retinopatia proliferante è caratterizzata da un’anomala formazione di neovasi che cresce sulla superficie del vitreo e si estende nella sua cavità.
In casi avanzati ciò può provocare un distacco di retina trazionale mentre dai neovasi possono originare emorragie vitreali. Questa forma di retinopatia ha diagnosi molto infausta perché il 70% degli occhi colpiti e non trattati diventa cieco entro cinque anni dai primi sintomi.
La maculopatia essudativa, invece è meno grave della precedente anche se può ugualmente condurre ad una notevole riduzione dell’acuità visiva.
La fotocoagulazione panretinica può diminuire la retinopatia proliferante e la neovascolarizzazione iridea. La fotocoagulazione precoce riduce il rischio di sviluppo del glaucoma neovascolare. In molti casi di emorragie vitreali può essere utile la vitrectomia.
Patologia retinica tipica dei bulbi affette da miopia elevata. La gravità delle alterazioni retiniche aumenta all’aumentare del difetto refrattivo ed è causata dal maggiore allungamento dell’occhio miopico rispetto alla norma (26 mm).
Ciò provoca una serie di alterazioni anatomiche e la retina subisce degli stiramenti o delle lesioni periferiche come piccoli fori perché non riesce ad adattarsi bene allungamento del bulbo, pertanto il tessuto retinico rimane meno irrorato dai vasi sanguigni.
Questa condizione provoca l’assottigliamento di uno strato della retina (epitelio pigmentato) che va incontro a piccole rotture. Nei casi gravi si arriva alla scomparsa dell’epitelio pigmentato retinico e la presenza dei fori provoca il distacco della membrana retinica.
Questa patologia inizia in età giovanile e può progredire nel tempo. Non esiste una terapia medica per la retinopatia miopica ma si possono curare le sue complicazioni (rotture, fori e aree di maggior assottigliamento retinico).
La retinopatia miopica può comportare una minorazione visiva rilevante a causa delle sue complicazioni come la neovascolarizzazione coroideale, (CNV) che può avere un’evoluzione lenta e dopo un brusco peggioramento si può assistere ad un miglioramento dell’acuità visiva dovuto alla riduzione dei fenomeni emorragici ed essudativi.
Quando la CNV va incontro a cicatrizzazione si può formare uno scotoma che con il tempo può allargarsi, con conseguente diminuzione della vista sino alla cecità legale.
Altra causa di minorazione visiva può essere lo stafiloma miopico, generalmente presente in giovane età, la cui gravità può aumentare negli anni: quando lo stafiloma è centrale, infatti, nel 50% dei casi il paziente diventa legalmente cieco solo dopo i 60 anni.
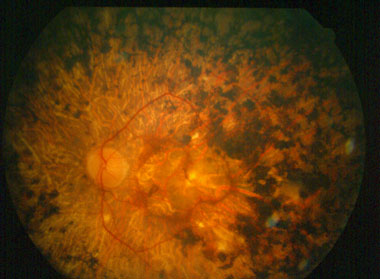
Gruppo di patologie che si manifestano con cecità notturna e riduzione concentrica del campo visivo periferico, sintomi associati a fotofobia ed abbagliamento. La patologia è bilaterale, lentamente progressiva e colpisce i bastoncelli provocando un deficit visivo notturno che può diventare sintomatico già nella prima infanzia. Si tratta di un gruppo di patologie prettamente oculari ma alcune forme rare hanno importanti associazioni sistemiche.
La retinite puramente oculare presenta ereditarietà ben definita nel 50% dei casi con dominanza recessiva legata al sesso. Si manifesta con uno scotoma anulare nella media periferia della retina che si estende gradualmente, tanto da ridurre ed alla fine impedire la visione centrale, consentendo solo quella periferica.
Le manifestazioni successive possono comprendere opacità degenerative del vitreo, cataratta e miopia. Si può associare ad ipoacusia congenita.
Ad oggi, nessuna terapia è in grado di arrestare l’evoluzione della degenerazione retinica pigmentosa.
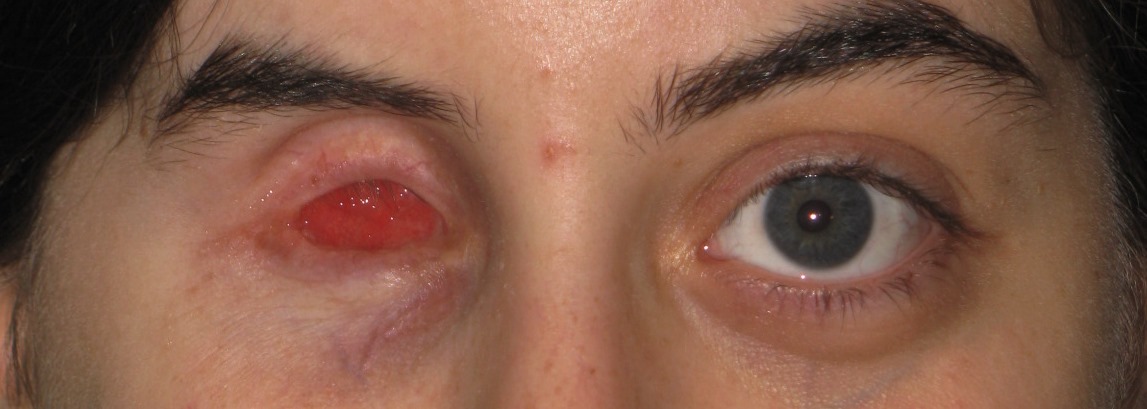
La cavità anoftalmica adatta ad accogliere la protesi oculare deve avere i seguenti requisiti:
Quando non esistono questi presupposti si ha a che fare con una cavità contratta, che presenta cioè un deficit relativo o assoluto per l’accoglimento della protesi. Ciò può accadere nei casi in cui la perdita del bulbo sia secondaria ad un trauma, all’uso di protesi inadatte che hanno leso progressivamente i tessuti, agli effetti della radioterapia o a patologie che hanno compromesso la qualità e la funzionalità dei tessuti orbitari o degli annessi.
Il trattamento chirurgico della cavità contratta è uno dei più difficili problemi della chirurgia ricostruttiva e si basa generalmente sul posizionamento di un innesto che sostituisca il tessuto andato perduto. Qualsiasi tipo di innesto venga utilizzato, è indispensabile che il protesista sia in grado di realizzare una protesi su misura subito dopo l’intervento, non appena il conformatore post-operatorio viene rimosso. La presenza della protesi e l’azione dinamica della chiusura della palpebra sopra di essa, prevengono infatti la contrazione dell’innesto aumentando le possibilità di riuscita dell’intervento.

Riduzione della sensibilità retinica (scotoma relativo) o sua scomparsa (scotoma assoluto) per cui le immagini non si percepiscono più in alcune aree della retina. In particolare, in presenza di uno scotoma assoluto ogni percezione visiva è perduta, mentre con uno scotoma relativo si perde la percezione cromatica (alcuni o tutti i colori ad eccezione del bianco).
Lo scotoma è chiamato negativo quando non si percepiscono, del tutto o in parte, gli oggetti fissati a causa di una macchia scura. Spesso non viene notato dal paziente se non interferisce con la visione centrale. Al contrario è detto positivo quando si percepisce una macchia a luminosità intermittente e di colore variabile che si proietta sugli oggetti fissati.
Gli scotomi negativi possono anche derivare da una patologia del nervo ottico (glaucoma, neurite ottica o neuropatia ottica ischemica). Uno scotoma bilaterale è di solito dovuto ad una lesione delle vie ottiche.
Se le lesioni sono permanenti (come lesioni delle vie ottiche e del tessuto retinico) non esiste una terapia risolutiva: quando la causa riguarda le cellule nervose, ad esempio nel caso di glaucoma e otticopatie, generalmente la capacità visiva dell’area interessata è definitivamente compromessa.
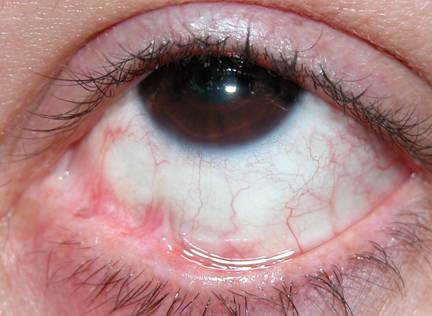
Aderenze tra la congiuntiva tarsale e la congiuntiva bulbare di natura congenita o più frequentemente, conseguente ad eventi accidentali (traumi o causticazioni) o patologie della superficie oculare.
E opportuno applicare un apposito conformatore che evitando il contatto tra le parti, favorisca la ricrescita della congiuntiva palpebrale. La congiuntiva bulbare, invece, può essere ripristinata solo chirurgicamente.
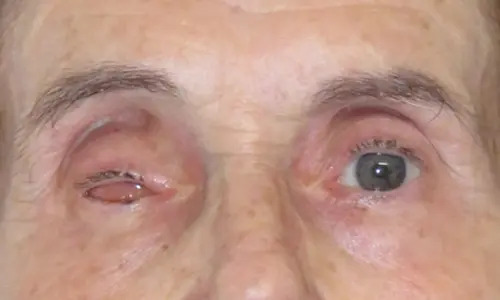
La sindrome dell’occhio fantasma può essere descritta come la percezione dell’area oculare amputata, e può essere associata al dolore e può essere causata da un’alterazione del sistema somatosensoriale o da un danno ai neuroni centrali o periferici.
Sebbene l’occhio abbia un’importante innervazione somatosensoriale le percezioni fantasma non sono frequenti ed è presente in circa un terzo dei pazienti anoftalmici. Le allucinazioni visive sono solitamente elementari e sono correlate a vite passate. Rasmussen et al hanno riportato questo fenomeno nel 36% dei pazienti anoftalmici studiati, ma solo l’1% di questi pazienti ha sperimentato allucinazioni visive complesse.
Le sensazioni fantasma possono essere descritte come parestesia, disestesia o iperpatia; i pazienti possono avvertire prurito periorbitale, percepire la presenza delle palpebre e immaginare di poter vedere con entrambi gli occhi.
Diversi fattori potrebbero agire da innesco, come la fatica, lo stress o una diversa illuminazione. Diversi studi hanno mostrato la somiglianza tra gli inneschi e gli alleviatori del dolore fantasma dell’arto amputato e quelli dell’occhio amputato, e la durata del dolore prima della rimozione dell’occhio e il rischio di sviluppare successivamente dolore fantasma sembrano essere correlati.
Diversi farmaci possono essere utilizzati per il trattamento, come antidepressivi, anticonvulsivanti, antagonisti del recettore N-metil-D-aspartato e oppioidi. Il supporto psicologico è essenziale, poiché tutti questi pazienti hanno bisogno di ridurre l’ansia per migliorare la loro qualità di vita.
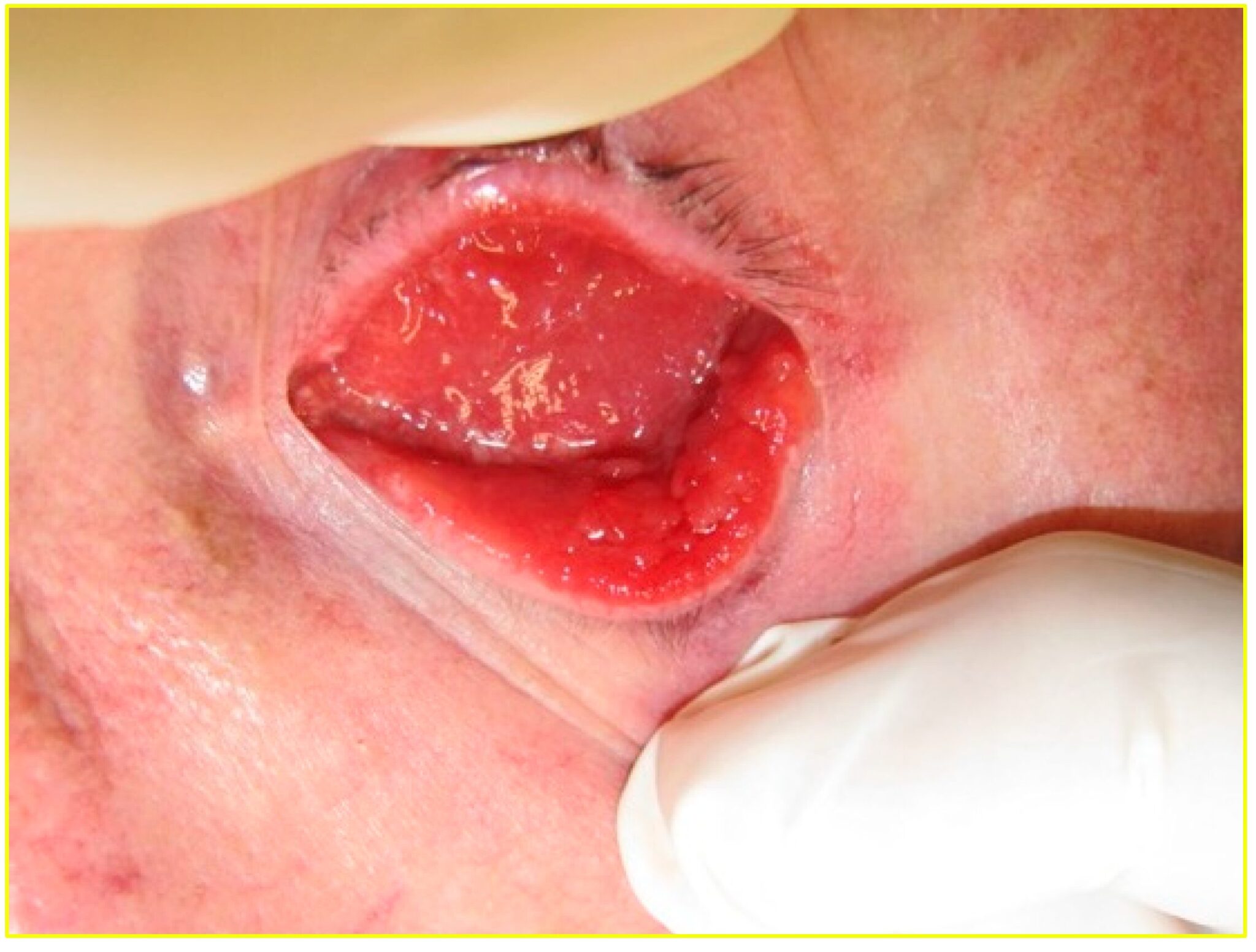
La sindrome dell’occhio fantasma può essere descritta come la percezione dell’area oculare amputata, e può essere associata al dolore e può essere causata da un’alterazione del sistema somatosensoriale o da un danno ai neuroni centrali o periferici.
Sebbene l’occhio abbia un’importante innervazione somatosensoriale le percezioni fantasma non sono frequenti ed è presente in circa un terzo dei pazienti anoftalmici. Le allucinazioni visive sono solitamente elementari e sono correlate a vite passate. Rasmussen et al hanno riportato questo fenomeno nel 36% dei pazienti anoftalmici studiati, ma solo l’1% di questi pazienti ha sperimentato allucinazioni visive complesse.
Le sensazioni fantasma possono essere descritte come parestesia, disestesia o iperpatia; i pazienti possono avvertire prurito periorbitale, percepire la presenza delle palpebre e immaginare di poter vedere con entrambi gli occhi.
Diversi fattori potrebbero agire da innesco, come la fatica, lo stress o una diversa illuminazione. Diversi studi hanno mostrato la somiglianza tra gli inneschi e gli alleviatori del
La sindrome dell’orbita anoftalmica secca (DASS) è una condizione comune che si verifica principalmente dopo l’enucleazione o l’eviscerazione. A causa di un’insufficiente lubrificazione e dell’instabilità del film lacrimale della superficie congiuntivale, si verificano livelli crescenti di disagio, secrezione frequente e intolleranza alla protesi.
Nel tempo, questi cambiamenti portano a secchezza della superficie, secrezione mucosa e infiammazione congiuntivale cronica, che possono ridurre il comfort della protesi.
La DASS è una condizione multifattoriale dovuta ad alterazioni anatomiche e fisiologiche conseguenti alla rimozione del bulbo oculare:
E’ un’alterazione del meccanismo che regola la secrezione e la distribuzione del film lacrimale che colpisce in maggioranza le persone tra i 40 e i 60 anni con maggiore incidenza di sesso femminile (probabilmente a causa degli squilibri ormonali che avvengono durante la menopausa).
Quando si alterano la quantità o la qualità delle lacrime la superficie corneale tende a seccarsi poiché si riduce o viene a mancare del tutto il film lacrimale e la superficie oculare esterna non risulta più lubrificata.
I sintomi tipici sono bruciore, sensazione di corpo estraneo, arrossamento oculare e fotofobia, al risveglio anche difficoltà di apertura delle palpebre e annebbiamento visivo.
Le cause possono essere patologiche come blefariti e congiuntiviti (anche allergiche) ed anche deficit di vitamina A (ipovitaminosi) che provoca la diminuzione del numero di cellule caliciformi che producono lo strato mucoso del film lacrimale.
Un’altra causa può essere l’alterazione della dinamica palpebrale (ammiccamento) nel caso di paresi del muscolo facciale, nell’esoftalmo da ipertiroidismo o in seguito a traumi.
L’occhio secco può essere anche sintomo di una patologia sistemica autoimmune come l’artrite reumatoide, il lupus eritematoso sistemico, la sclerodermia o la sindrome di Sjögren.
Altri fattori di rischio sono lo smog, il fumo di sigaretta, l’esposizione eccessiva all’aria condizionata o all’aria calda e l’utilizzo prolungato del videoterminale.
I sintomi di questo disturbo sono attenuati da terapie che prevedono la protezione della superficie oculare mediante lacrime artificiali o gel lubrificanti. Ovviamente la riduzione dei sintomi avviene soltanto per breve tempo e quindi la terapia è prettamente palliativa.
Il gel oculare però ha un’azione umettante più prolungata e rimanendo a contatto più a lungo con la superficie oculare, soprattutto durante la notte e grazie alle sue proprietà fisiche, si lega alle lacrime formando un film lubrificante particolarmente resistente.
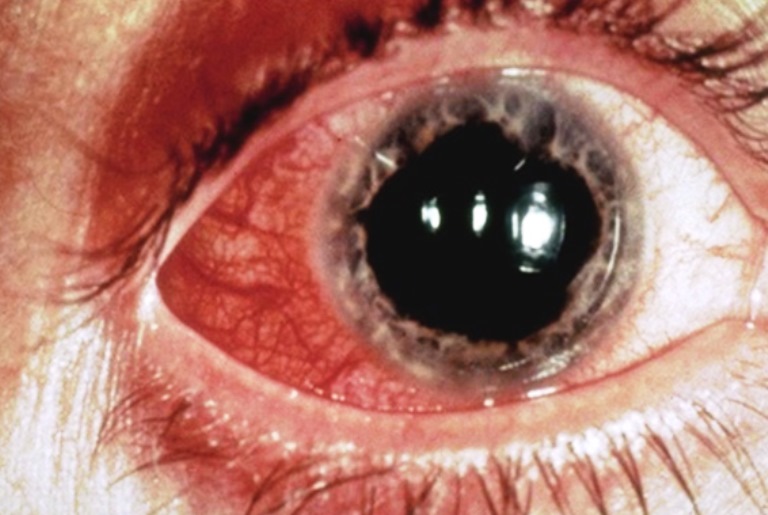
E’ una patologia rara, cronica, caratterizzata da una infiammazione dei vasi sanguigni in tutto il corpo. Oltre a causare ulcere orali e genitali ricorrenti e lesioni oculari, può causare anche vari tipi di lesioni cutanee, artrite, tromboflebiti, infiammazione dell’intestino e del sistema nervoso centrale. La causa comune di queste lesioni è una infiammazione dei vasi arteriosi e venosi.
Le cause della malattia non sono note. Non è una malattia infettiva o contagiosa, né la trasmissione avviene per via sessuale ed è caratterizzata negli anni da periodi di remissione e riacutizzazione. I sintomi possono durare alcuni giorni o settimane, o possono persistere per mesi o anni. Provoca diversi livelli di disabilità che interferiscono con la qualità della vita.
L’apparato oculare è colpito dalla formazione di uveite anteriore e/o posteriore con l’ineteressamento retinico (vasculite) ed è la lesione più grave poiché determina la formazione di cicatrici retiniche. I sintomi sono quelli tipici delle iridocicliti acute, cioè perdita o annebbiamento della vista e dolore all’interno dell’occhio.
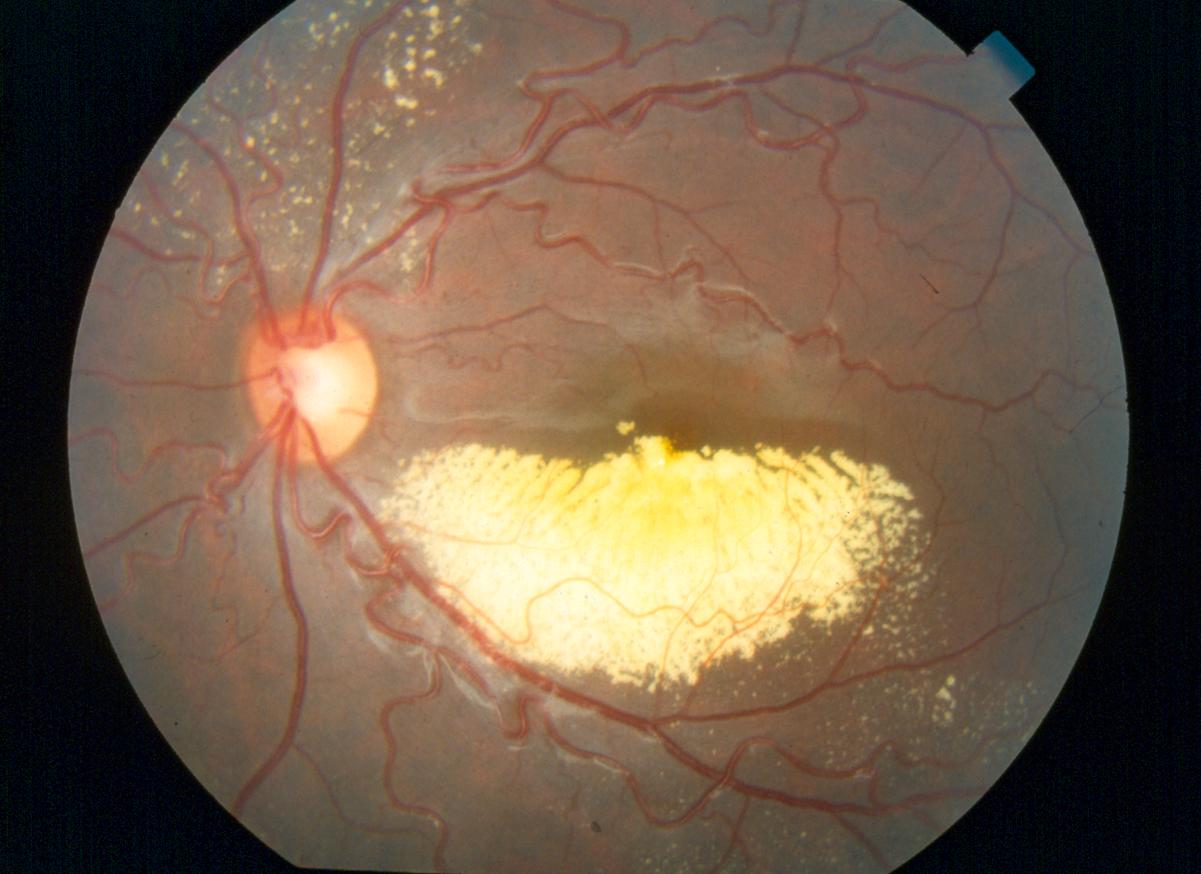
E’ una sindrome molto rara (un caso su ogni 30.000 nascite), di natura idiopatica, non ereditaria, caratterizzata da uno sviluppo abnorme dei vasi sanguigni della retina (telangiectasie) e dalla presenza di essudati intra e sotto retinei. Generalmente è monolaterale e colpisce prevalentemente i bambini di sesso maschile, con un picco di incidenza compreso tra 1 e 8 anni.
La patologia è caratterizzata dall’associazione di amiotrofia facio-scapolo-omerale, teleangectasie retiniche e sordità mono o bilaterale di tipo sensoriale. La miopatia esordisce nell’infanzia e progredisce rapidamente fino alla perdita della deambulazione. Negli stadi avanzati, la malattia provoca distacco totale della retina, leucocoria, glaucoma doloroso secondario ad angolo chiuso ed è difficilmente differenziabile dal retinoblastoma.
Anche nota come displasia oculo-auricolo-vertebrale o microsomia emifacciale, è una sindrome polimalformativa caratterizzata da anomalie oculari, microtia uni o bilaterale, ipoplasia mandibolare, microsomia emifacciale, anomalie del cavo orale, anomalie dell’apparato respiratorio, anomalie genito-urinarie, anomalie cardiache, anomalie del sistema nervoso, anomalie scheletriche vertebrali e anomalie cranio-facciali.
Ha una frequenza variabile da 1/3500 a 1/26000 nati ed interessa più spesso l’emifaccia dx (dx/sx=3/2) ed il sesso maschile (m/f=3/2). L’eziologia è sconosciuta sono stati proposti fattori genetici e ambientali come agenti causali. Sebbene nella maggior parte dei casi sia sporadica, sono state descritte sia le modalità di trasmissione autosomica recessiva sia quella autosomica dominante.
Le manifestazioni tipiche della sindrome sono:
Sottosviluppo dei tessuti di una parte del viso, della mandibola inferiore e superiore. Il mento tende ad inclinarsi verso quel lato. Il tessuto della guancia è spesso sottosviluppato e unitamente al problema mandibolare crea un’asimmetria del viso. Anomalia del condilo e conseguenti problemi dentali.
Residui cartilaginei nella sede preauricolare e assenza o malformazione del padiglione auricolare, presenza di appendici pre-auricolari, stenosi-atresia del condotto uditivo, sordità trasmissiva o neurosensoriale, deficit dell’udito per lesione sia dell’orecchio medio che interno.
Cisti dermoide epibulbare, dermoide sub congiuntivale o orbitale anteriore, coloboma della palpebra superiore, blefaroptosi, anomalia del sistema lacrimale, tumori epibulbari, micro-anoftalmia, anomalia della retina, strabismo.
emispondili ed emivertebre a livello lombare, toracico dorsale e cervicale, scoliosi, occipitalizzazione dell’atlante, spina bifida. Malformazioni del cranio dalla parte colpita, microrbitismo, zigomo ed osso temporale più piccoli.
macrostomia, fistole tracheo-esofagee, atresia esofagea, malformazioni strutturali e funzionali di faringe e laringe che possono contribuire ad incrementare il rischio di ostruzione delle vie aeree, di menomazione della comunicazione e di mobilità. Labbro leporino, labio palato schisi, palato ogivale. La sindrome può causare una grave apnea ostruttiva del sonno che se non trattata causa ritardo di crescita e mentale.
Anomalie renali e genito-urinarie (ectopia del testicolo, criptorchidismo), anomalie cardiache (Tetralogia di Fallot, difetto del setto interventricolare, costrizione dell’aorta), anomalie del sistema nervoso e cerebrali (ipoplasia del ponte encefalico, agenesia del corpo calloso, idrocefalo, microcefalo, encefalocele, nel 5%-15% dei casi ritardo mentale), anomalie dell’apparato respiratorio (ad alcuni bambini è stata praticata tracheotomia poco dopo la nascita. In alcuni casi la mandibola era così malformata che la lingua ostruiva il respiro durante il sonno).
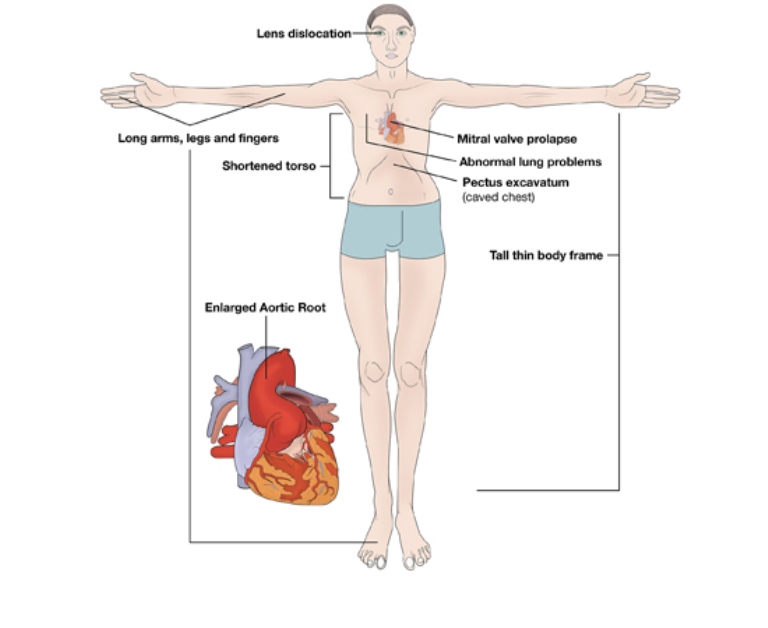
E’ una grave condizione medica classificata come disturbo ereditario del tessuto connettivo che in primo luogo colpisce le ossa ed i legamenti (sistema scheletrico), gli occhi, i polmoni ma soprattutto il cuore ed il sistema cardiovascolare che presenta i rischi maggiori per la sopravvivenza del paziente.
Le ossa e i legamenti vengono colpiti in diverse maniere. Un individuo affetto sarà alto, magro e dalle articolazioni sciolte e flessibili. Braccia, gambe e dita possono essere sproporzionatamente lunghe in confronto al tronco. La flessibilità si estende ai piedi che spesso sono piatti. La curvatura spinale è diffusa e può divenire abbastanza seria se non trattata. Le ossa del torace possono o avere una protuberanza (detta comunemente torace carenato) o incavato (pectus incavatum) dovuto ad un sovrasviluppo delle costole. Il palato è generalmente molto arcuato ed i denti sono storti. Il volto può apparire lungo e stretto in confronto con la forma generale del corpo. 1 bambini hanno spesso uno sguardo profondo e sembrano più grandi rispetto al loro fratelli e sorelle della stessa età non affetti.
Il cristallino dislocato (ectopia lentis) in circa il 75% degli affetti dalla sindrome. La dislocazione dei cristallini è per questo un importante segnale che la sindrome di Marfan è presente. Generalmente la dislocazione del cristallino appare sin dalla prima infanzia e sembra mai oltre gli 11 anni.
Un altra disfunzione comune a livello oculare è la miopia. L’ indebolimento della vista può essere blando o abbastanza grave ed indipendente dalla presenza di dislocamento del cristallino. Fori o lacerazioni della retina sono infrequenti e così pure il rischio di distacco.
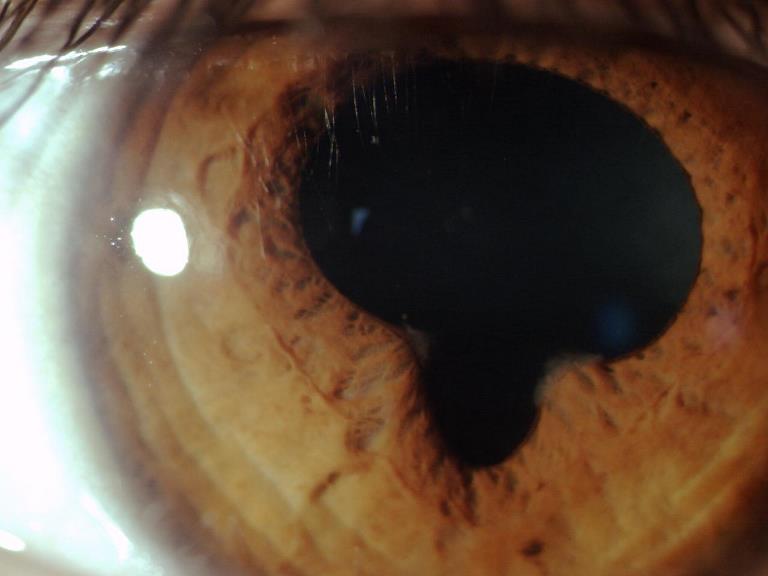
Aderenze che si formano tra l’iride e le altre strutture della camera anteriore dell’occhio in seguito a processi infiammatori che coinvolgono l’iride. Le sinechie anteriori si evidenziano tra l’iride e la superficie posteriore della cornea, provocano spesso la formazione di opacità corneali nella zona di contatto tra le due superfici e occlusione dell’angolo camerulare, con conseguente aumento della pressione oculare.
Le sinechie inoltre possono portare alla deformazione del forame pupillare che può presentarsi stirato in varie direzioni e generare disturbi visivi e abbagliamento.
Se queste ultime si estendono a 360° si può determinare un’occlusione del forame pupillare che impedisce all’umore acqueo di essere drenato correttamente, provocando quindi l’insorgenza di un glaucoma acuto secondario.
Le sinechie posteriori invece si localizzano tra la parte posteriore dell’iride e la faccia anteriore del cristallino, provocando spesso cataratta o glaucoma da occlusione della pupilla.
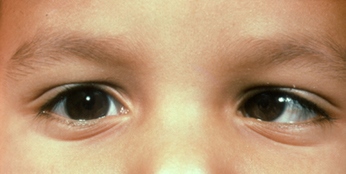
Patologia che consiste nella condizione in cui gli assi visivi dei due occhi non sono allineati. Ciò può comportare diplopia se insorge in età adulta, quando invece è congenito il cervello del bambino esclude spontaneamente l’occhio deviato proprio per evitare la visione doppia: l’occhio non utilizzato in questo caso si definisce “ambliope”.
In base alla direzione della deviazione dell’orientamento lo strabismo può essere convergente (verso l’interno), divergente (verso l’esterno) o verticale (verso l’alto).
Lo strabismo convergente può essere di tipo:
Lo strabismo divergente, molto meno frequente e riscontrabile soprattutto nel bambino in età scolare e nell’adulto. A differenza di quello convergente si manifesta spesso in modo intermittente e varia secondo che il soggetto guardi lontano o vicino. Può essere:
Lo strabismo verticale, invece, è generalmente associato ad una deviazione orizzontale ed è spesso causato da paralisi di origine ostetrica o neonatale.
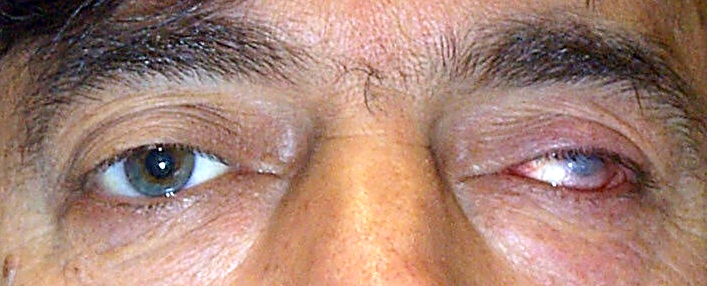
Riduzione patologica del volume del bulbo causata dall’interruzione del flusso ematico o dalla perdita di sostanza interna. Il bulbo può diventare atrofico in seguito a postumi di un trauma, di una ferita perforante o in seguito a patologie come emorragia retinica, panoftalmite od altri processi infettivi. L’occhio subatrofico è quasi sempre non vedente ma spesso non è in quiete e provoca dolore ed infiammazione continua.
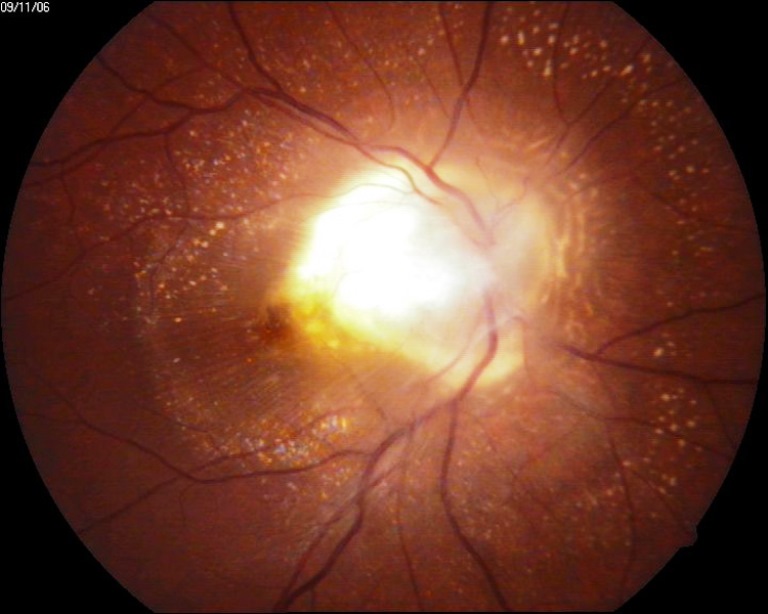
Patologia causata dalla toxocara, un nematode parassita che risiede nel piccolo intestino di cani, gatti e carnivori selvatici e he produce ogni giorno centinaia di uova dove gli embrioni maturano e raggiungono lo stadio larvale infettivo. La prevalenza delle uova di Toxocara nel suolo è correlata al numero di cani presenti in quella zona ed a fattori climatici del luogo.
L’infezione umana è dovuta all’accidentale ingestione di uova infette e la trasmissione può avvenire mediante l’ingestione di cibo contaminato o di terra, per cui i bambini di età inferiore ai 10 anni sono più soggetti ad entrambi i tipi di infezione per la vicinanza con il terreno e per il più frequente contatto con cuccioli di cane.
L’infezione umana può presentarsi in maniera asintomatica, simile ad un’infezione virale e l’incidenza della toxocariasi oculare è anch’essa più frequente nei bambini e costituisce l’1-2% delle uveiti in questo gruppo d’età.
La Toxocara può essere considerata come un possibile agente causale delle uveiti posteriori e di quelle diffuse infantili, l’età media dei pazienti alla diagnosi di toxocariasi oculare è infatti di 7,5 anni e l’80% dei pazienti presentano un’età inferiore ai 16 anni.
Possono essere riscontrate tre differenti manifestazioni oculari: l’endoftalmite cronica, il granuloma posteriore e il granuloma periferico.
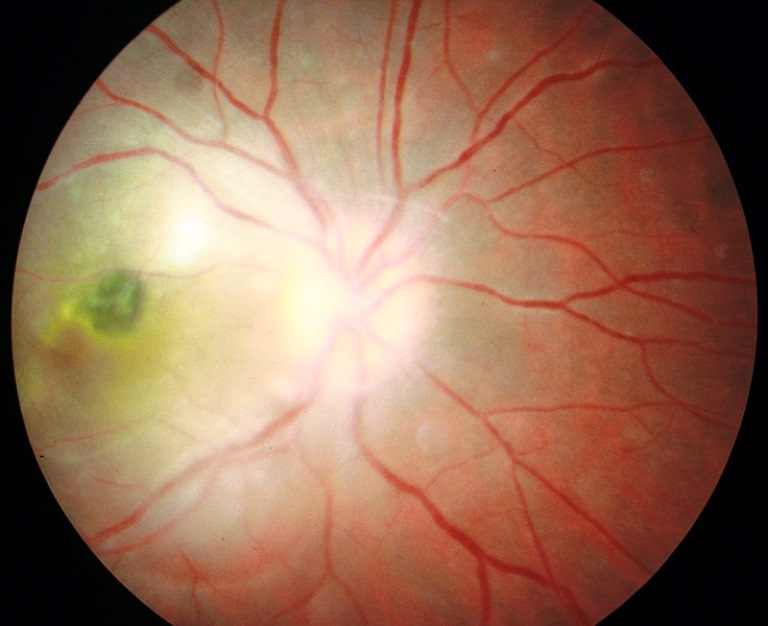
Patologia spesso congenita causata dal protozoo Toxoplasma Gondii presente nelle feci degli uccelli e dei mammiferi come il gatto, nei rettili, nei molluschi ma rilevabile anche nella carne cruda e nelle verdure non lavate accuratamente. Il parassita predilige il tessuto nervoso e può attraversare la placenta durante l’infezione della madre, provocando in determinate circostanze malformazioni o addirittura l’aborto o la morte in utero.
La toxoplasmosi primaria può essere complicata da sintomi gravi come la corioretinite oltre a sintomi attribuibili a una malattia autoimmune. Quest’ultima eventualità è frequente nei malati di Aids o nei soggetti trapiantati, per i quali spesso l’evoluzione è drammatica perché la risposta alla terapia è insufficiente.
La toxoplasmosi acquisita è un’infezione primaria contratta sia con l’ingestione di ovocisti tramite cibo (carni bovine e suinem crude o non sufficientemente cotte, verdure crude non lavate), sia più raramente per ingestione a seguito del contatto con oggetti, terreno o altro materiale contaminato dalle ovocisti di toxoplasma.
L’infezione decorre generalmente in forma asintomatica o con sintomi lievi caratterizzati da interessamento linfoghiandolare. L’interessamento oftalmico della toxoplasmosi provoca l’uveite posteriore infettiva con possibili formazioni di cicatrici a livello retinico ma l’infezione può causare anche gravi patologie come calcificazioni endocraniche, idrocefalo, ritardo mentale ed epilessia.
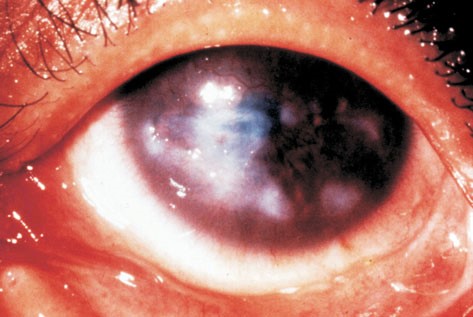
Grave patologia oculare dovuta all’infezione provocata dal batterio Chlamydia Tracomatis. I primi sintomi si manifestano con arrossamento, fotofobia, forte bruciore, lacrimazione ed edema palpebrale. Segno tipico di questa patologia è la palpebra rivolta verso l’interno che si salda alla congiuntiva (entropion cicatriziale) e con essa la trichiasi delle ciglia ed il lagoftalmo, cui conseguono cicatrici corneali fino alla formazione del panno corneale.
L’infezione si trasmette attraverso secrezioni oculari infette oppure indirettamente in seguito a contatto con materiale contaminato ma anche le mosche che si nutrono di secrezioni oculari possono trasmettere la malattia.
Il tracoma è la principale causa di cecità tra le malattie contagiose ed è endemico in alcune zone dell’Africa, del Medio Oriente, dell’India e del Sud-Est asiatico, prediligendo i bambini nei primi due anni di vitae di conseguenza le donne, giustificando l’incidenza due o tre volte più alta della forma più grave cicatriziale nel sesso femminile rispetto a quello maschile.
Patologia delle ciglia che deviano dal loro percorso naturale e crescono in direzione del bulbo, venendo a contatto con la cornea e abradendola. Spesso associata all’entropion, si riscontra anche nelle patologie palpebrali croniche ed in seguito a traumi od esiti cicatriziali. La trichiasi che nella sua fase matura è molto dolorosa, deve essere corretta chirurgicamente perché a lungo andare può causare danni corneali irreversibili.
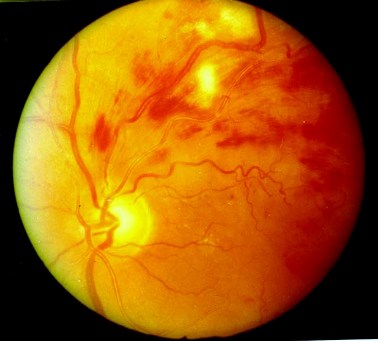
Patologia vascolare che consiste nell’occlusione dell’arteria centrale retinica e che può provocare in pochi minuti lesioni irreversibili alla funzione visiva. L’occlusione può essere dovuta a embolia (placche arterosclerotiche disseminate, endocardite, emboli grassosi) oppure a trombosi di un’arteria centrale sclerotica.
La trombosi retinica può avere origine da un’occlusione venosa o arteriosa:
Occlusione Venosa (Più comune): A causa di un trombo il sangue non riesce a defluire correttamente, provocando edema ed emorragie. Il danno dipende dall’estensione del blocco e dal livello di ischemia.
Occlusione Arteriosa (Più grave): Un embolo blocca l’arrivo del sangue nell’arteria e causa un’ischemia totale e una perdita della vista quasi istantanea; se non trattata entro pochi minuti o nelle prime ore può determinare un infarto retinico da cui può conseguire la cecità permanente, determinando anche frequentemente la tisi del bulbo che in seguito al mancato afflusso sanguigno entra in una condizione di ipotonia e si atrofizza.
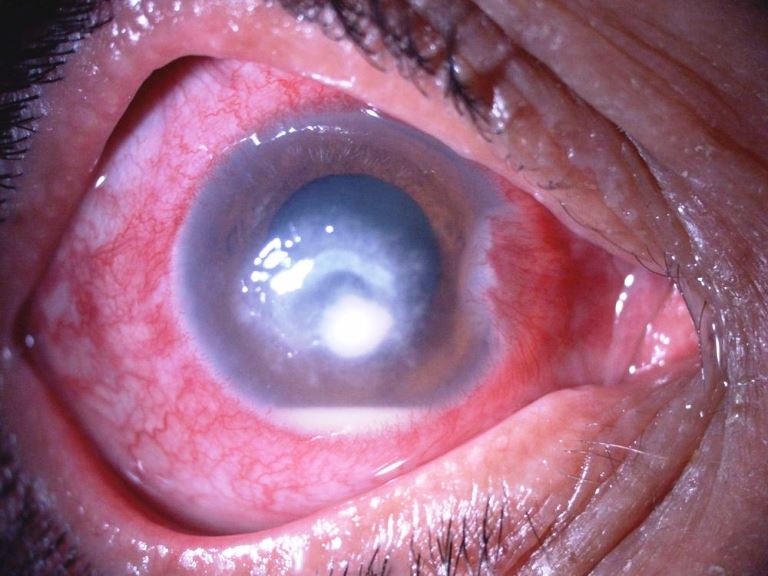
Ulcera batterica
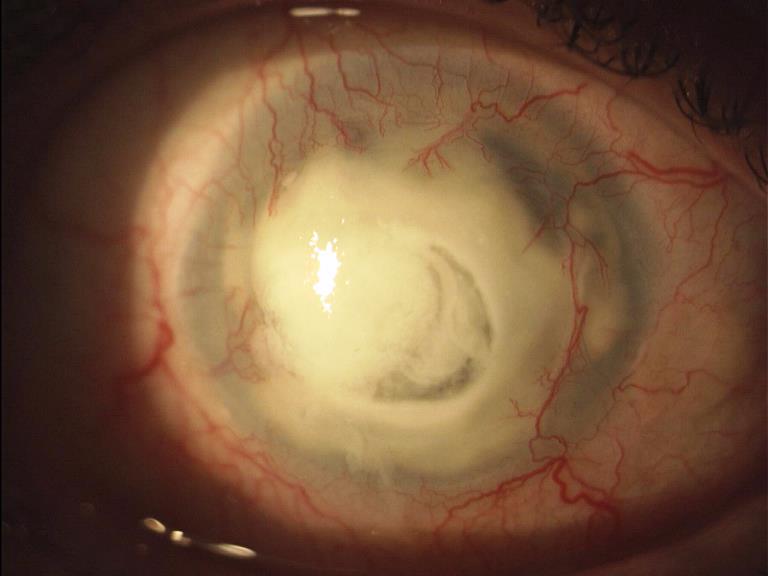
Neovasi cicatriziali
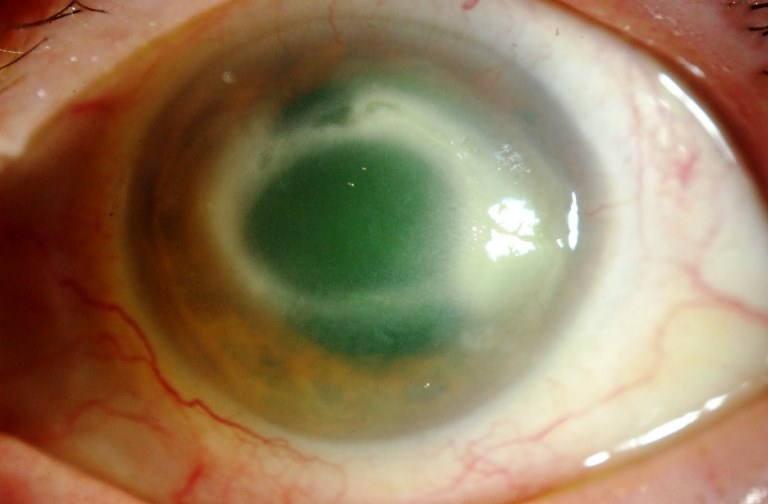
Ulcera infetta
Lesione della cornea in seguito ad un processo infettivo: assume l’aspetto di un piccolo cratere con necrosi locale del tessuto corneale, dovuta all’invasione da parte di batteri, funghi, virus o Acanthamoeba. Se trascurata può mettere in pericolo le altre strutture oculari.
L’ulcera corneale batterica è provocata più frequentemente da Stafilococco, Pseudomonas, o Streptococcus pneumoniae. L’infezione è causata spesso dall’uso delle lenti a contatto durante il sonno, dalla loro inadeguata sterilizzazione ma anche da un trauma corneale oppure da un corpo estraneo corneale.
L’ulcera corneale può anche essere la complicanza di una cheratite neurotrofica, blefarite cronica, congiuntivite (specialmente batterica), tracoma, cheratopatia bollosa e pemfigoide cicatriziale. Può essere anche la conseguenza di alterazioni del trofismo corneale secondarie a carenza di vitamina A o di malnutrizione proteica ed ancora da anomalie palpebrali come l’entropion, la trichiasi, il lagoftalmo e l’esoftalmo).
L’ulcera causate da funghi che sono più croniche rispetto a quelle batteriche, si infiltrano profondamente e possono presentare alla periferia distinte zone di infiltrazione.
Le ulcere corneali provocate da Acanthamoeba, invece, sono molto dolorose e possono manifestarsi con difetti transitori dell’epitelio corneale, multipli infiltrati stromali e successivamente con un unico infiltrato opaco anulare.
L’ulcera erpetica (o dendritica) è provocata dal virus Herpes Simplex o Varicella Zoster in soggetti che hanno subito una prima infezione precedentemente con o senza manifestazioni di herpes labiale o palpebrale. È di norma monolaterale, ha tendenza recidivante e può anche essere concomitante a rash cutaneo di origine erpetica. Questo tipo di ulcera associata a cheratite da herpes simplex è molto pericolosa perché può essere particolarmente refrattaria a qualsiasi trattamento terapeutico.
I sintomi tipici dell’ulcera corneale sono il dolore, senso di corpo estraneo, fotofobia e lacrimazione ma inizialmente possono essere minimi e facilmente sottovalutati. L’ulcera corneale si manifesta inizialmente come opacità superficiale grigiastra e circoscritta e in seguito va incontro a necrosi e suppurazione provocando un’ulcerazione escavata. È presente un difetto dell’epitelio corneale superficiale che si colora di verde con fluoresceina. È frequente un’accentuata iperemia perilimbare e nei casi cronici dal limbus si formano dei neovasi che si dirigono verso l’ulcerazione (neovascolarizzazione corneale).
L’ulcera si può estendere fino a coinvolgere tutta la superficie corneale o può penetrare in profondità nella camera anteriore dove può comparire un ipopion.
Le complicanze sono in diretta relazione con la profondità dell’ulcera: più questa è profonda, più sono gravi. Le ulcere corneali guariscono con formazione di tessuto cicatriziale che causa opacizzazione della cornea e diminuzione del visus. Se il trattamento è assente o tardivo possono formarsi complicazioni molto gravi come la perforazione corneale con prolasso dell’iride e la panoftalmite.
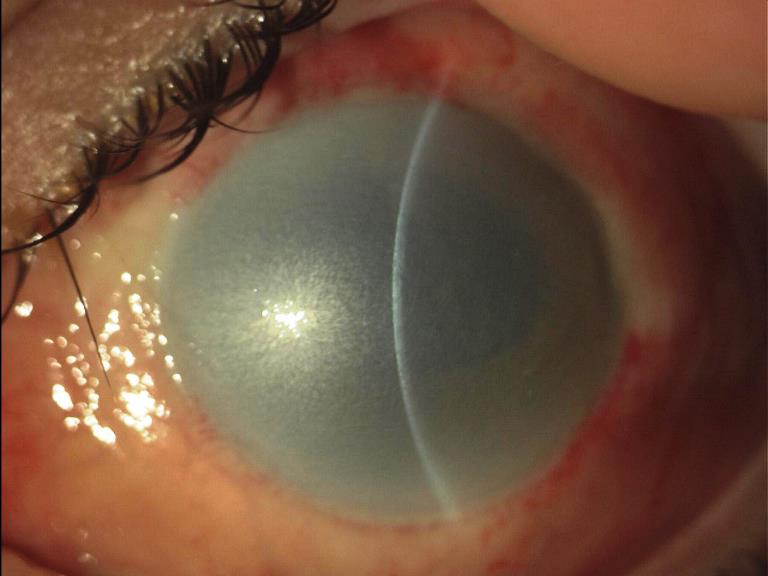
Le causticazioni o ustioni chimiche provocano gravi reazioni tossiche del bulbo. Quelle associate ad un’alterazione del PH possono indurre danni permanenti come risultato del contatto con l’agente chimico.
Le lesioni dovute ad ammoniaca ed altre sostanze alcaline sono le più gravi per la loro rapida diffusione endobulbare che spesso in pochi minuti causano danni oculari permanenti come ulcere profonde con perdita di epitelio corneale e successiva necrosi della congiuntiva.
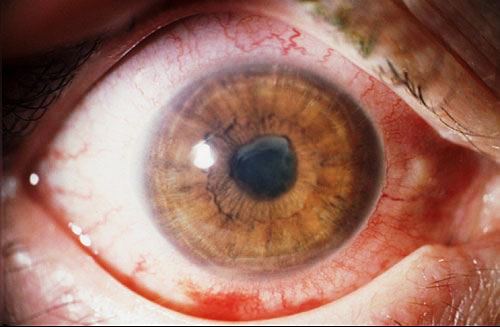
Patologia oculare dovuta ad un processo infiammatorio che interessa l’uvea, membrana costituita da iride, corpo ciliare e coroide. In rapporto alla zona del bulbo dove ha origine l’uveite può essere:
In base al decorso clinico viene classificata in uveite acuta o cronica e in base all’eziologia in uveite esogena (provocate da agenti provenienti dall’esterno) o endogena(da agenti già presenti nel nostro organismo). Può essere inoltre causata da virus, batteri, funghi e parassiti (uveite infettiva) o anche in forma non infettiva in associazione ad altre patologie sistemiche.
I sintomi dipendono dalla sede colpita dall’infiammazione:
Complicazioni:
Le forme anteriori si possono complicare con la formazione di sinechie tra l’iride e la superficie anteriore del cristallino e quando si formano attorno alla pupilla possono provocare il mancato passaggio di umore acqueo dalla camera posteriore del bulbo oculare a quella anteriore, con conseguente aumento della pressione oculare. Altre complicazioni dell’uveite anteriore sono la cataratta e le sofferenze corneali.
L’uveite intermedia ha come complicazioni l’edema maculare cistoide ed il distacco di retina per trazione.
Nell’uveite posteriore le complicazioni riguardano il coinvolgimento della macula da parte dell’infiammazione che provoca edema maculare, fenomeni di occlusione dei vasi della retina, formazione di neovasi coroideali e infiammazione del nervo ottico.
L’uveite è una patologia insidiosa che va trattata con tempestività poiché le forme più gravi possono portare alla cecità.
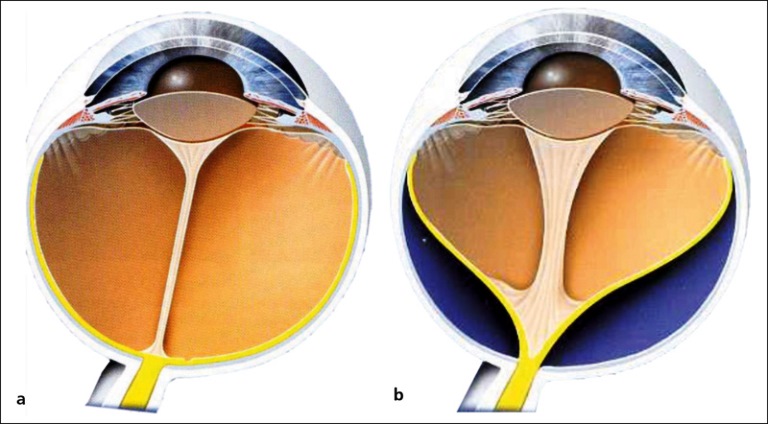
Bulbo normale (a) e con Vitreo primitivo iperplastico (b)
La persistenza del Vitreo primitivo iperplastico è una rara patologia congenita caratterizzata dalla mancata regressione del vitreo primario e della vascolarizzazione vitreale normalmente presente solo nel feto. Può essere distinto in forma anteriore, posteriore e completa a seconda delle strutture oculari coinvolte.
La forma anteriore corrisponde a tessuto fibrovascolare adeso al cristallino e ai processi ciliari. Quella posteriore consiste in un cordone fibrovascolare vitreale che emerge dal nervo ottico e decorre anteriormente. Nella forma completa i due quadri si associano.
Ciò accade perché nella crescita prenatale il segmento posteriore dell’occhio è ricco di rami dell’arteria ialoide proveniente dal nervo ottico (vitreo primario). Successivamente questi vasi sanguigni scompaiono per essere sostituiti dal vitreo secondario che a sua volta origina il vitreo trasparente e avascolare tipico di ogni occhio correttamente formato.
Se alcuni vasi sanguigni permangono tipicamente nell’arteria ialoide che porta dal disco ottico alla superficie posteriore del cristallino, permane anche il vitreo primario.
L’occhio con vitreo iperplastico primitivo persistente è frequente nei nati prematuri affetti da Retinopatia del Pretermine (R.O.P.), ed è di solito più piccolo del controlaterale normale (microftalmo o ipoplasia). Molto frequentemente questa patologia si associa a distacco di retina, cataratta e glaucoma, tutti nella forma congenita.
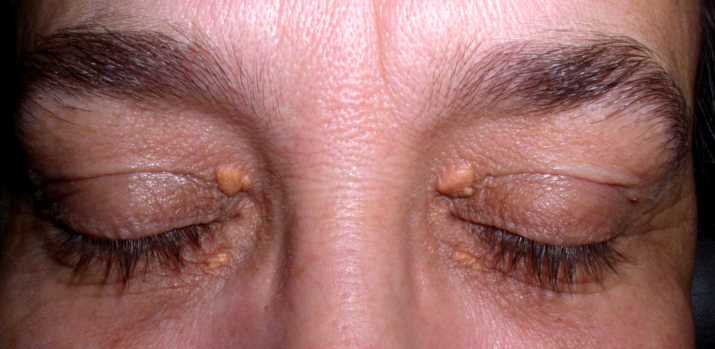
E’ un accumulo di grassi, in particolare di colesterolo localizzato sulle palpebre. Particolarmente diffuso dopo i 40 anni, colpisce di frequente anche i giovani e si presenta come una placca molle e giallastra di forma e dimensione variabile che sporge sulla palpebra. Non causa dolore e non interferisce con la vista. Lo xantelasma può essere rimosso chirurgicamente ma spesso si riforma spontaneamente.

Patologia del bulbo causata da carenza di vitamina A, il cui sintomo caratteristico è la sindrome dell’occhio secco. Può degenerare in ulcera corneale e cheratomalacia.
E’ la maggiore causa di cecità infantile nei Paesi in via di sviluppo. Si stima che 250 milioni di bambini in età prescolare soffrano di carenza di Vitamina A, ogni anno 350.000 bambini diventino ciechi e sempre ogni anno muoiano 2 milioni di bambini per questa patologia.
E’ dovuta alla perdita delle cellule congiuntivali caliciformi e mucosecernenti e ad un’alterazione delle cellule epiteliali congiuntivali. Queste alterazioni provocano la cheratinizzazione dell’epitelio congiuntivale che diviene secco (xerosi congiuntivale). La xerosi si manifesta a livello della congiuntiva bulbare con mancanza di umidità della mucosa, suo ispessimento e raggrinzimento, perdita della pigmentazione e della trasparenza.
La secchezza corneale e congiuntivale può portare a ulcerazione e cheratomalacia con una conseguente perdita della vista nel 50% dei pazienti non trattati. L’ulcerazione corneale sembra essere dovuta al trauma e alla mancanza di proteine associata a una carenza di vitamina A.
Copyright© 2019. Tutti i diritti sono riservati.
Sono vietate le riproduzioni delle immagini e del testo, anche parziali.