






Raro tumore maligno pediatrico delle parti molli in età pediatrica che rappresenta circa il 3-4% di tutti i tumori infantili. Il nome è formato dalla combinazione di tre parole più piccole: “rabdo”, che significa a forma di bacchetta, “mio” che sta per muscolo, e “sarcoma”. Infatti le cellule del rabdomiosarcoma hanno caratteristiche che ricordano le cellule muscolari dell’embrione.
Colpisce i bambini da 1 a 5 anni ma anche gli adolescenti e più raramente ne possono essere affetti anche gli adulti. Può nascere in qualsiasi parte del corpo, ma generalmente è più frequente a livello di testa-collo (40%), apparato genitourinario (20%) , arti e tronco (20%).
La causa di questo tumore non è nota ma un certo numero di bambini con rabdomiosarcoma ha anche malformazioni congenite di altri organi (cuore, intestino, cervello, etc.) o rare malattie genetiche, come la neurofibromatosi di tipo I.
In generale si rivela come una massa in crescita più o meno rapida, spesso compare una tumefazione o di un nodulo. I tumori che insorgono nella regione della testa e/o del collo possono causare mal di testa, nausea, vomito, diplopia, paralisi facciale e ostruzione delle vie aeree.
E’ distinto in due sottotipi, quello embrionale e quello alveolare.
Il rabdomiosarcoma embrionale è il più frequente e tende a svilupparsi a livello della testa e del collo, della vescica, della vagina nelle femmine e attorno alla prostata e ai testicoli nel maschio.
In presenza di un rabdomiosarcoma localizzato (cioè senza metastasi) le possibilità di guarigione arrivano fino al 70% ma in circa il 20-25% dei casi Il rabdomiosarcoma si presenta con metastasi già al momento della diagnosi. Gli organi più frequentemente colpiti da metastasi sono i polmoni, le ossa, il midollo osseo, i tessuti sottocutanei.

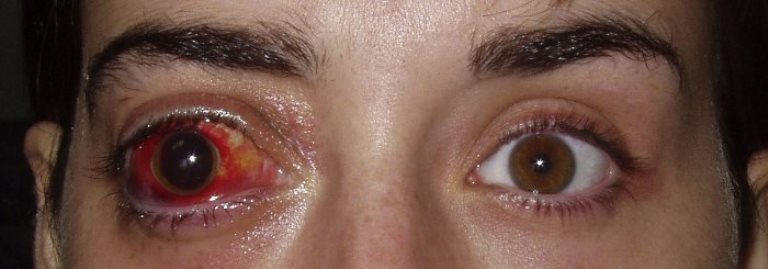
Tumore maligno della retina monolaterale o bilaterale che generalmente colpisce i bambini sotto i 6 anni ma più comunemente diagnosticato tra uno e due anni. Sebbene si tratti di un tumore raro (1 caso su 20.000 nascite) è una delle patologie maligne più comuni dell’infanzia.
Può essere monolaterale o bilaterale (nel 35% dei casi) e in questo caso è sempre presente un’ereditarietà genetica.
Cause: compare quando una cellula della retina in crescita sviluppa una mutazione, inducendola a crescere fuori controllo e diventare cancerosa. A volte è presente in diversi membri della famiglia del bambino colpito. Se la mutazione accade in famiglia, vi è un 50% di probabilità che ciò accada anche ai bambini. Il tumore può diffondersi agli occhi attraverso il nervo ottico fino a raggiungere anche cervello, polmoni e ossa.
Sintomi: si presentano generalmente entro i primi 5 anni di vita ed il segno principale è un riflesso biancastro del fondo oculare, detto leucocoria, dovuto alla massa tumorale retinica che occupa il corpo vitreo, correlata spesso a deviazione degli assi oculari (dovuta alla perdita di fissazione dell’occhio affetto).
Altri sintomi caratteristici sono: pupilla distorta, riflesso bianco nelle foto con il flash, strabismo, arrossamento e dolore all’occhio, abbassamento del visus ed iride di colore diverso in ciascun occhio.
Prognosi: Se la lesione non è diffusa oltre il bulbo quasi tutti i pazienti possono essere curati con discrete possibilità di guarigione: è questo infatti uno dei pochi tumori che occasionalmente possono regredire con la terapia. Questa può richiedere però un trattamento aggressivo e nel caso peggiore la rimozione del bulbo. Se la lesione è infiltrata oltre il bulbo, infatti, le probabilità di successo della terapia sono molto minori.
Terapia: esistono dei protocolli terapeutici che impongono determinati trattamenti in base allo stadio di malattia. I trattamenti locali includono: laser fotocoagulazione, crioterapia, termoterapia transpupillare e brachiterapia (applicazione di placche radioattive).
A questi trattamenti viene associata attualmente la chemioterapia per via sistemica. Negli ultimi tempi si sta affermando l’utilizzo di chemioterapia per via arteriosa, attraverso l’arteria oftalmica.
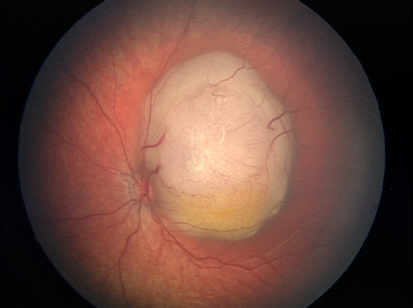
Retinite massiva caratterizzata da un anomalo sviluppo dei vasi sanguigni della retina (telangectasia), con progressiva deposizione di essudati intraretinici e sottoretinici che portano potenzialmente al distacco essudativo della retina.
La patologia è spesso monolaterale e può complicarsi con l’insorgere di un distacco di retina, un glaucoma secondario o una cataratta complicata. Si presenta prevalentemente in bambini di sesso maschile verso la fine della prima decade di vita, non è ereditaria ed il suo decorso clinico è variabile ma quasi sempre progressivo.
La retinopatia di Coats può associarsi ad altre malformazioni vascolari come angiomi facciali, sindrome di Alport, retinopatia pigmentosa ed altre.
Stadiazione:
Fase 1, caratterizzata solo da teleangectasie
Fase 2, presenta teleangectasie ed essudazione con coinvolgimento della fovea
Fase 3, totale o parziale distacco della retina
Fase 4, totale distacco della retina con glaucoma
Fase 5, cecità, occhio indolore o doloroso, spesso con cataratta ed eventuale tisi tubulare
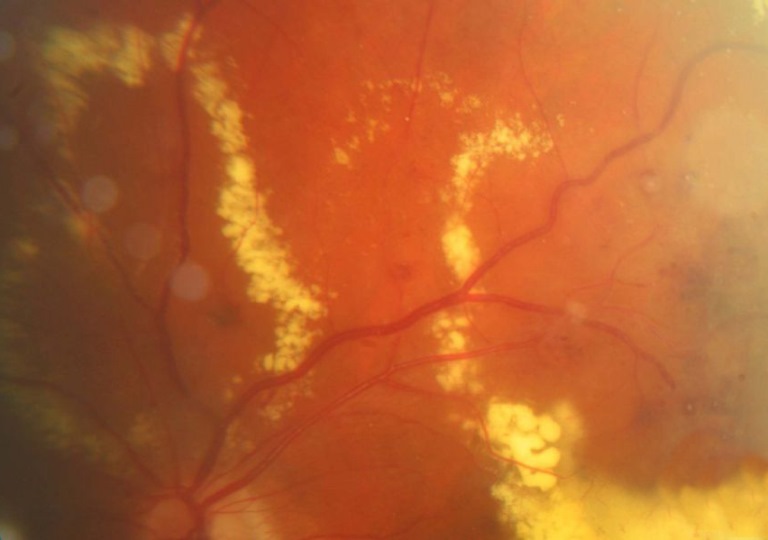
Patologia della retina definita anche fibroplasia retrolentale, spesso bilaterale che si manifesta nei nati prematuri con rischio maggiore quanto minore è il tempo di gestazione e quanto è più basso il peso alla nascita.
La vascolarizzazione retinica fetale origina dalla papilla e finché questa non è completa rimane sensibile alle elevate concentrazioni di ossigeno tipiche del trattamento in incubatrice.
La patologia provoca la formazione di neovasi nella periferia retinica e successivamente emorragia vitreale, fibrosi e trazione retinica che nello stadio più avanzato si trasforma in fibroplasia retrolentale. La sua evoluzione può portare alla cecità in seguito a distacco per trazione.
Allo stadio iniziale la terapia consiste nel criotrattamento o nel trattamento laser, “bruciando” la retina periferica per evitare che possa fornire degli stimoli per la formazione dei nuovi vasi dannosi. Quando si manifestano gli ultimi stadi della malattia è necessario intervenire chirurgicamente con vitrectomia o piombaggio sclerale. La R.O.P. è una delle maggiori cause di ftisi del bulbo nei bambini.
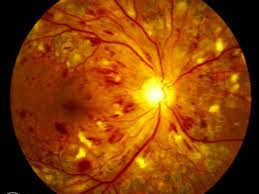
Questa patologia, importante causa di cecità, può essere particolarmente grave nei pazienti affetti da diabete mellito insulino-dipendente (DID) di tipo 1. Si manifesta spesso anche nel diabete mellito cronico non insulino-dipendente (DNID) di tipo 2. Il grado della retinopatia sembra strettamente correlato con la durata del diabete.
Il diabete colpisce il microcircolo retinico anche se nelle prime fasi della malattia non si evidenziano grosse alterazioni dei vasi. I primi segni della retinopatia diabetica sono spesso rappresentati dalle dilatazioni venose e da piccole macchie rosse (microaneurismi capillari) che si osservano all’esame oftalmoscopico nel polo retinico posteriore.
Le emorragie retiniche, l’edema profondo e gli essudati lipidici possono alterare la funzione maculare. I sintomi tardivi sono una diminuzione generalizzata del visus provocata da edema maculare.
L’edema maculare è una causa comune di alterazione visiva nei diabetici e si manifesta con la comparsa di essudati molli (microinfarti causati da ridotta perfusione retinica) ed essudati duri che invece sono provocati da un edema cronico.
La retinopatia proliferante è caratterizzata da un’anomala formazione di neovasi che cresce sulla superficie del vitreo e si estende nella sua cavità.
In casi avanzati ciò può provocare un distacco di retina trazionale mentre dai neovasi possono originare emorragie vitreali. Questa forma di retinopatia ha diagnosi molto infausta perché il 70% degli occhi colpiti e non trattati diventa cieco entro cinque anni dai primi sintomi.
La maculopatia essudativa, invece è meno grave della precedente anche se può ugualmente condurre ad una notevole riduzione dell’acuità visiva.
La fotocoagulazione panretinica può diminuire la retinopatia proliferante e la neovascolarizzazione iridea. La fotocoagulazione precoce riduce il rischio di sviluppo del glaucoma neovascolare. In molti casi di emorragie vitreali può essere utile la vitrectomia.
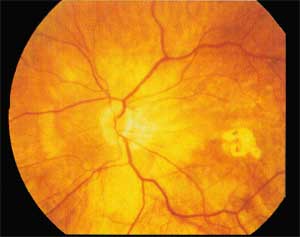
Patologia retinica tipica dei bulbi affette da miopia elevata. La gravità delle alterazioni retiniche aumenta all’aumentare del difetto refrattivo ed è causata dal maggiore allungamento dell’occhio miopico rispetto alla norma (26 mm).
Ciò provoca una serie di alterazioni anatomiche e la retina subisce degli stiramenti o delle lesioni periferiche come piccoli fori perché non riesce ad adattarsi bene allungamento del bulbo, pertanto il tessuto retinico rimane meno irrorato dai vasi sanguigni.
Questa condizione provoca l’assottigliamento di uno strato della retina (epitelio pigmentato) che va incontro a piccole rotture. Nei casi gravi si arriva alla scomparsa dell’epitelio pigmentato retinico e la presenza dei fori provoca il distacco della membrana retinica.
Questa patologia inizia in età giovanile e può progredire nel tempo. Non esiste una terapia medica per la retinopatia miopica ma si possono curare le sue complicazioni (rotture, fori e aree di maggior assottigliamento retinico).
La retinopatia miopica può comportare una minorazione visiva rilevante a causa delle sue complicazioni come la neovascolarizzazione coroideale, (CNV) che può avere un’evoluzione lenta e dopo un brusco peggioramento si può assistere ad un miglioramento dell’acuità visiva dovuto alla riduzione dei fenomeni emorragici ed essudativi.
Quando la CNV va incontro a cicatrizzazione si può formare uno scotoma che con il tempo può allargarsi, con conseguente diminuzione della vista sino alla cecità legale.
Altra causa di minorazione visiva può essere lo stafiloma miopico, generalmente presente in giovane età, la cui gravità può aumentare negli anni: quando lo stafiloma è centrale, infatti, nel 50% dei casi il paziente diventa legalmente cieco solo dopo i 60 anni.
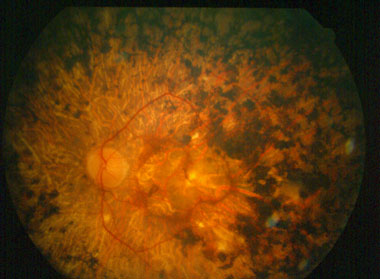
Gruppo di patologie che si manifestano con cecità notturna e riduzione concentrica del campo visivo periferico, sintomi associati a fotofobia ed abbagliamento. La patologia è bilaterale, lentamente progressiva e colpisce i bastoncelli provocando un deficit visivo notturno che può diventare sintomatico già nella prima infanzia. Si tratta di un gruppo di patologie prettamente oculari ma alcune forme rare hanno importanti associazioni sistemiche.
La retinite puramente oculare presenta ereditarietà ben definita nel 50% dei casi con dominanza recessiva legata al sesso. Si manifesta con uno scotoma anulare nella media periferia della retina che si estende gradualmente, tanto da ridurre ed alla fine impedire la visione centrale, consentendo solo quella periferica.
Le manifestazioni successive possono comprendere opacità degenerative del vitreo, cataratta e miopia. Si può associare ad ipoacusia congenita.
Ad oggi, nessuna terapia è in grado di arrestare l’evoluzione della degenerazione retinica pigmentosa.
La cavità anoftalmica adatta ad accogliere la protesi oculare deve avere i seguenti requisiti:
Quando non esistono questi presupposti si ha a che fare con una cavità contratta, che presenta cioè un deficit relativo o assoluto per l’accoglimento della protesi. Ciò può accadere nei casi in cui la perdita del bulbo sia secondaria ad un trauma, all’uso di protesi inadatte che hanno leso progressivamente i tessuti, agli effetti della radioterapia o a patologie che hanno compromesso la qualità e la funzionalità dei tessuti orbitari o degli annessi.
Il trattamento chirurgico della cavità contratta è uno dei più difficili problemi della chirurgia ricostruttiva e si basa generalmente sul posizionamento di un innesto che sostituisca il tessuto andato perduto. Qualsiasi tipo di innesto venga utilizzato, è indispensabile che il protesista sia in grado di realizzare una protesi su misura subito dopo l’intervento, non appena il conformatore post-operatorio viene rimosso. La presenza della protesi e l’azione dinamica della chiusura della palpebra sopra di essa, prevengono infatti la contrazione dell’innesto aumentando le possibilità di riuscita dell’intervento.

Riduzione della sensibilità retinica (scotoma relativo) o sua scomparsa (scotoma assoluto) per cui le immagini non si percepiscono più in alcune aree della retina. In particolare, in presenza di uno scotoma assoluto ogni percezione visiva è perduta, mentre con uno scotoma relativo si perde la percezione cromatica (alcuni o tutti i colori ad eccezione del bianco).
Lo scotoma è chiamato negativo quando non si percepiscono, del tutto o in parte, gli oggetti fissati a causa di una macchia scura. Spesso non viene notato dal paziente se non interferisce con la visione centrale. Al contrario è detto positivo quando si percepisce una macchia a luminosità intermittente e di colore variabile che si proietta sugli oggetti fissati.
Gli scotomi negativi possono anche derivare da una patologia del nervo ottico (glaucoma, neurite ottica o neuropatia ottica ischemica). Uno scotoma bilaterale è di solito dovuto ad una lesione delle vie ottiche.
Se le lesioni sono permanenti (come lesioni delle vie ottiche e del tessuto retinico) non esiste una terapia risolutiva: quando la causa riguarda le cellule nervose, ad esempio nel caso di glaucoma e otticopatie, generalmente la capacità visiva dell’area interessata è definitivamente compromessa.
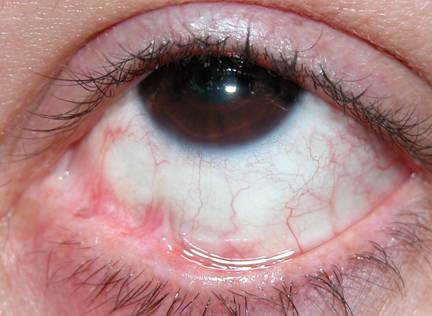
Aderenze tra la congiuntiva tarsale e la congiuntiva bulbare di natura congenita o più frequentemente, conseguente ad eventi accidentali (traumi o causticazioni) o patologie della superficie oculare.
E opportuno applicare un apposito conformatore che evitando il contatto tra le parti, favorisca la ricrescita della congiuntiva palpebrale. La congiuntiva bulbare, invece, può essere ripristinata solo chirurgicamente.
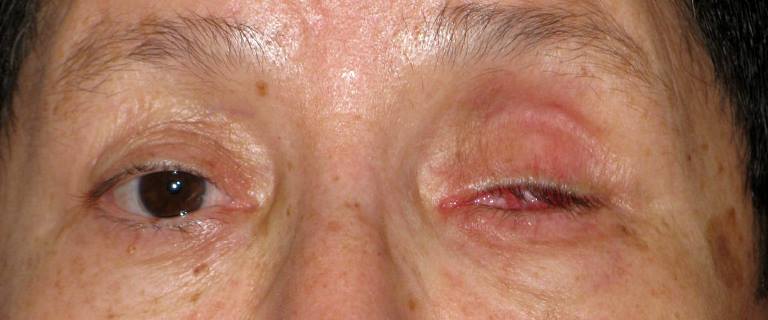
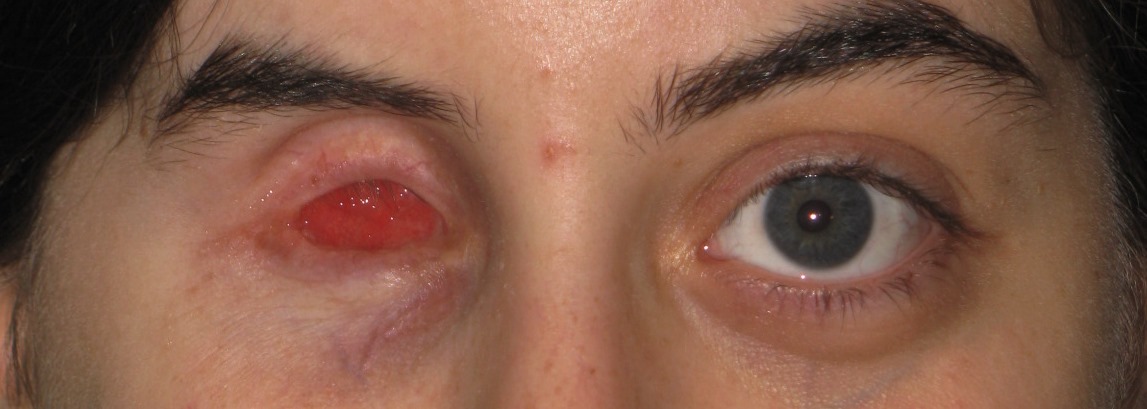
È un insieme di sintomi che possono esser presenti in pazienti che hanno subito l’enucleazione o eviscerazione del bulbo. In un significativo numero di pazienti si sviluppano le seguenti anomalie:
La sua patogenesi è complessa e si basa essenzialmente sul deficit di volume orbitario provocato essenzialmente dalla perdita del bulbo ed anche dal riassorbimento post-chirurgico del tessuto adiposo che riveste l’orbita.
È possibile ottenere buoni risultati estetici e funzionali con una chirurgia mirata a ripristinare il volume orbitario tramite innesto dermoadiposo od anche un semplice impianto acrilico nella capsula di Tenone. Con l’inserimento di un impianto orbitario al momento dell’intervento (impianto primario) o in un momento successivo (impianto secondario) si possono correggere in parte questi deficit estetici.
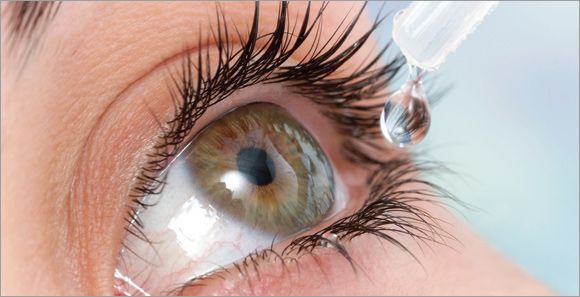
E’ un’alterazione del meccanismo che regola la secrezione e la distribuzione del film lacrimale che colpisce in maggioranza le persone tra i 40 e i 60 anni con maggiore incidenza di sesso femminile (probabilmente a causa degli squilibri ormonali che avvengono durante la menopausa).
Quando si alterano la quantità o la qualità delle lacrime la superficie corneale tende a seccarsi poiché si riduce o viene a mancare del tutto il film lacrimale e la superficie oculare esterna non risulta più lubrificata.
I sintomi tipici sono bruciore, sensazione di corpo estraneo, arrossamento oculare e fotofobia, al risveglio anche difficoltà di apertura delle palpebre e annebbiamento visivo.
Le cause possono essere patologiche come blefariti e congiuntiviti (anche allergiche) ed anche deficit di vitamina A (ipovitaminosi) che provoca la diminuzione del numero di cellule caliciformi che producono lo strato mucoso del film lacrimale.
Un’altra causa può essere l’alterazione della dinamica palpebrale (ammiccamento) nel caso di paresi del muscolo facciale, nell’esoftalmo da ipertiroidismo o in seguito a traumi.
L’occhio secco può essere anche sintomo di una patologia sistemica autoimmune come l’artrite reumatoide, il lupus eritematoso sistemico, la sclerodermia o la sindrome di Sjögren.
Altri fattori di rischio sono lo smog, il fumo di sigaretta, l’esposizione eccessiva all’aria condizionata o all’aria calda e l’utilizzo prolungato del videoterminale.
I sintomi di questo disturbo sono attenuati da terapie che prevedono la protezione della superficie oculare mediante lacrime artificiali o gel lubrificanti. Ovviamente la riduzione dei sintomi avviene soltanto per breve tempo e quindi la terapia è prettamente palliativa.
Il gel oculare però ha un’azione umettante più prolungata e rimanendo a contatto più a lungo con la superficie oculare, soprattutto durante la notte e grazie alle sue proprietà fisiche, si lega alle lacrime formando un film lubrificante particolarmente resistente.
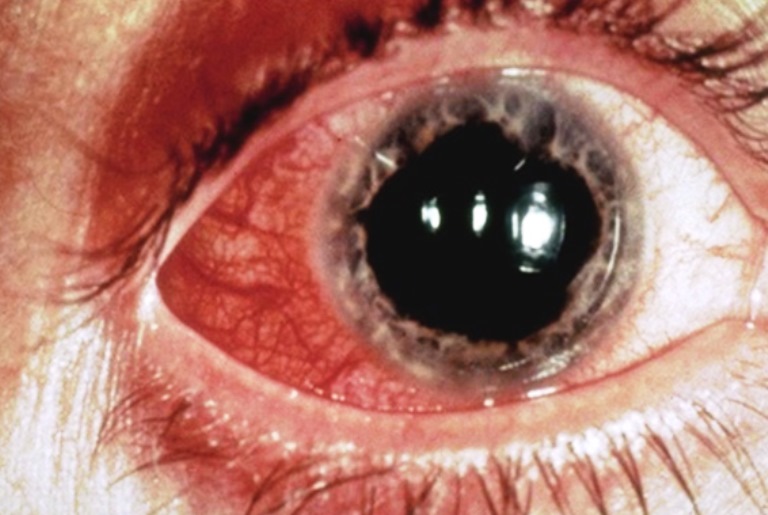
E’ una patologia rara, cronica, caratterizzata da una infiammazione dei vasi sanguigni in tutto il corpo. Oltre a causare ulcere orali e genitali ricorrenti e lesioni oculari, può causare anche vari tipi di lesioni cutanee, artrite, tromboflebiti, infiammazione dell’intestino e del sistema nervoso centrale. La causa comune di queste lesioni è una infiammazione dei vasi arteriosi e venosi.
Le cause della malattia non sono note. Non è una malattia infettiva o contagiosa, né la trasmissione avviene per via sessuale ed è caratterizzata negli anni da periodi di remissione e riacutizzazione. I sintomi possono durare alcuni giorni o settimane, o possono persistere per mesi o anni. Provoca diversi livelli di disabilità che interferiscono con la qualità della vita.
L’apparato oculare è colpito dalla formazione di uveite anteriore e/o posteriore con l’ineteressamento retinico (vasculite) ed è la lesione più grave poiché determina la formazione di cicatrici retiniche. I sintomi sono quelli tipici delle iridocicliti acute, cioè perdita o annebbiamento della vista e dolore all’interno dell’occhio.
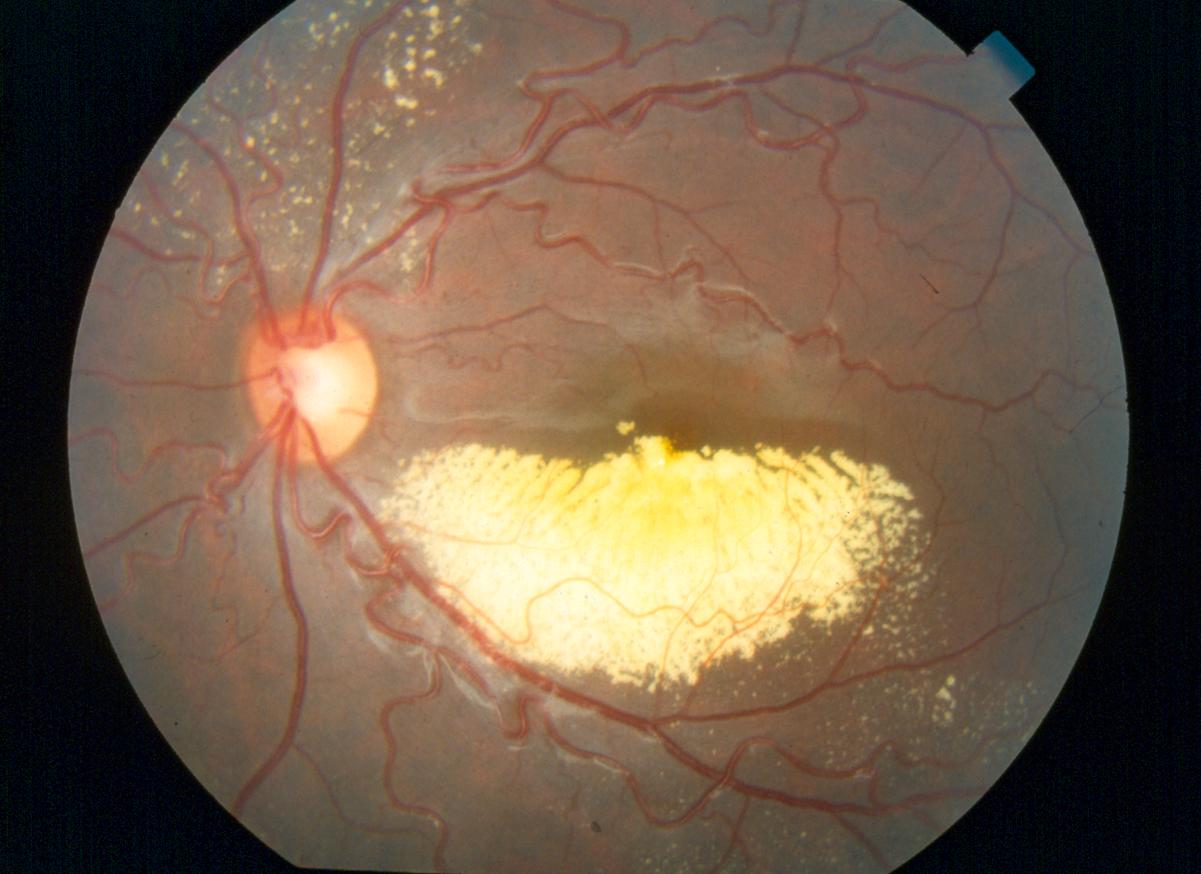
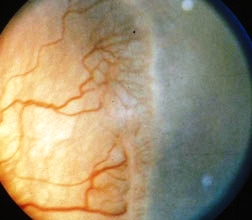
E’ una sindrome molto rara (un caso su ogni 30.000 nascite), di natura idiopatica, non ereditaria, caratterizzata da uno sviluppo abnorme dei vasi sanguigni della retina (telangiectasie) e dalla presenza di essudati intra e sotto retinei. Generalmente è monolaterale e colpisce prevalentemente i bambini di sesso maschile, con un picco di incidenza compreso tra 1 e 8 anni.
La patologia è caratterizzata dall’associazione di amiotrofia facio-scapolo-omerale, teleangectasie retiniche e sordità mono o bilaterale di tipo sensoriale. La miopatia esordisce nell’infanzia e progredisce rapidamente fino alla perdita della deambulazione. Negli stadi avanzati, la malattia provoca distacco totale della retina, leucocoria, glaucoma doloroso secondario ad angolo chiuso ed è difficilmente differenziabile dal retinoblastoma.
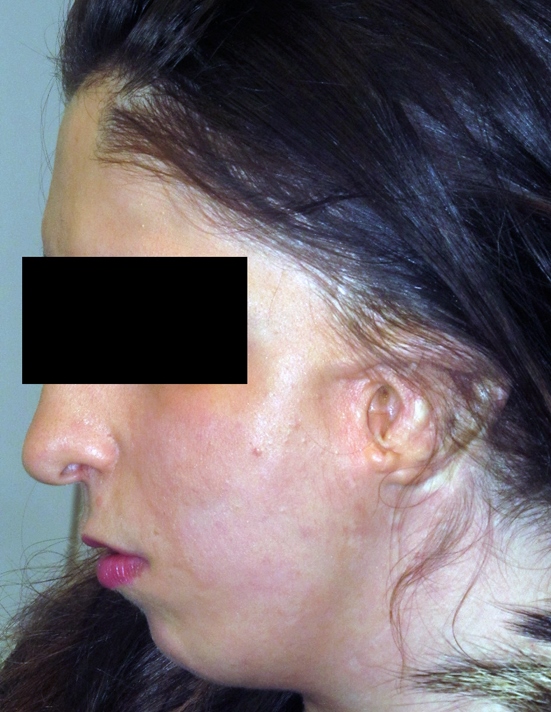
Anche nota come displasia oculo-auricolo-vertebrale o microsomia emifacciale, è una sindrome polimalformativa caratterizzata da anomalie oculari, microtia uni o bilaterale, ipoplasia mandibolare, microsomia emifacciale, anomalie del cavo orale, anomalie dell’apparato respiratorio, anomalie genito-urinarie, anomalie cardiache, anomalie del sistema nervoso, anomalie scheletriche vertebrali e anomalie cranio-facciali.
Ha una frequenza variabile da 1/3500 a 1/26000 nati ed interessa più spesso l’emifaccia dx (dx/sx=3/2) ed il sesso maschile (m/f=3/2). L’eziologia è sconosciuta sono stati proposti fattori genetici e ambientali come agenti causali. Sebbene nella maggior parte dei casi sia sporadica, sono state descritte sia le modalità di trasmissione autosomica recessiva sia quella autosomica dominante.
Le manifestazioni tipiche della sindrome sono:
Sottosviluppo dei tessuti di una parte del viso, della mandibola inferiore e superiore. Il mento tende ad inclinarsi verso quel lato. Il tessuto della guancia è spesso sottosviluppato e unitamente al problema mandibolare crea un’asimmetria del viso. Anomalia del condilo e conseguenti problemi dentali.
Residui cartilaginei nella sede preauricolare e assenza o malformazione del padiglione auricolare, presenza di appendici pre-auricolari, stenosi-atresia del condotto uditivo, sordità trasmissiva o neurosensoriale, deficit dell’udito per lesione sia dell’orecchio medio che interno.
Cisti dermoide epibulbare, dermoide sub congiuntivale o orbitale anteriore, coloboma della palpebra superiore, blefaroptosi, anomalia del sistema lacrimale, tumori epibulbari, micro-anoftalmia, anomalia della retina, strabismo.
emispondili ed emivertebre a livello lombare, toracico dorsale e cervicale, scoliosi, occipitalizzazione dell’atlante, spina bifida. Malformazioni del cranio dalla parte colpita, microrbitismo, zigomo ed osso temporale più piccoli.
macrostomia, fistole tracheo-esofagee, atresia esofagea, malformazioni strutturali e funzionali di faringe e laringe che possono contribuire ad incrementare il rischio di ostruzione delle vie aeree, di menomazione della comunicazione e di mobilità. Labbro leporino, labio palato schisi, palato ogivale. La sindrome può causare una grave apnea ostruttiva del sonno che se non trattata causa ritardo di crescita e mentale.
Anomalie renali e genito-urinarie (ectopia del testicolo, criptorchidismo), anomalie cardiache (Tetralogia di Fallot, difetto del setto interventricolare, costrizione dell’aorta), anomalie del sistema nervoso e cerebrali (ipoplasia del ponte encefalico, agenesia del corpo calloso, idrocefalo, microcefalo, encefalocele, nel 5%-15% dei casi ritardo mentale), anomalie dell’apparato respiratorio (ad alcuni bambini è stata praticata tracheotomia poco dopo la nascita. In alcuni casi la mandibola era così malformata che la lingua ostruiva il respiro durante il sonno).
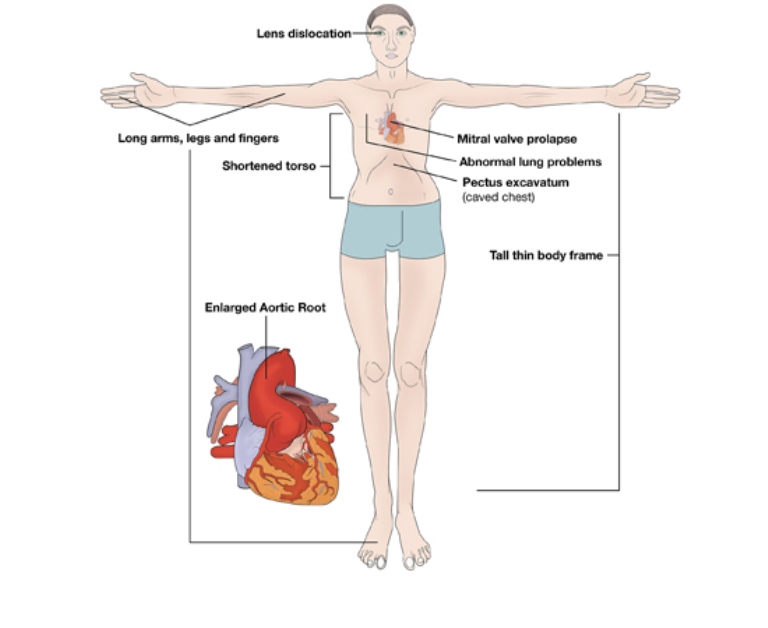
E’ una grave condizione medica classificata come disturbo ereditario del tessuto connettivo che in primo luogo colpisce le ossa ed i legamenti (sistema scheletrico), gli occhi, i polmoni ma soprattutto il cuore ed il sistema cardiovascolare che presenta i rischi maggiori per la sopravvivenza del paziente.
Le ossa e i legamenti vengono colpiti in diverse maniere. Un individuo affetto sarà alto, magro e dalle articolazioni sciolte e flessibili. Braccia, gambe e dita possono essere sproporzionatamente lunghe in confronto al tronco. La flessibilità si estende ai piedi che spesso sono piatti. La curvatura spinale è diffusa e può divenire abbastanza seria se non trattata. Le ossa del torace possono o avere una protuberanza (detta comunemente torace carenato) o incavato (pectus incavatum) dovuto ad un sovrasviluppo delle costole. Il palato è generalmente molto arcuato ed i denti sono storti. Il volto può apparire lungo e stretto in confronto con la forma generale del corpo. 1 bambini hanno spesso uno sguardo profondo e sembrano più grandi rispetto al loro fratelli e sorelle della stessa età non affetti.
Il cristallino dislocato (ectopia lentis) in circa il 75% degli affetti dalla sindrome. La dislocazione dei cristallini è per questo un importante segnale che la sindrome di Marfan è presente. Generalmente la dislocazione del cristallino appare sin dalla prima infanzia e sembra mai oltre gli 11 anni.
Un altra disfunzione comune a livello oculare è la miopia. L’ indebolimento della vista può essere blando o abbastanza grave ed indipendente dalla presenza di dislocamento del cristallino. Fori o lacerazioni della retina sono infrequenti e così pure il rischio di distacco.
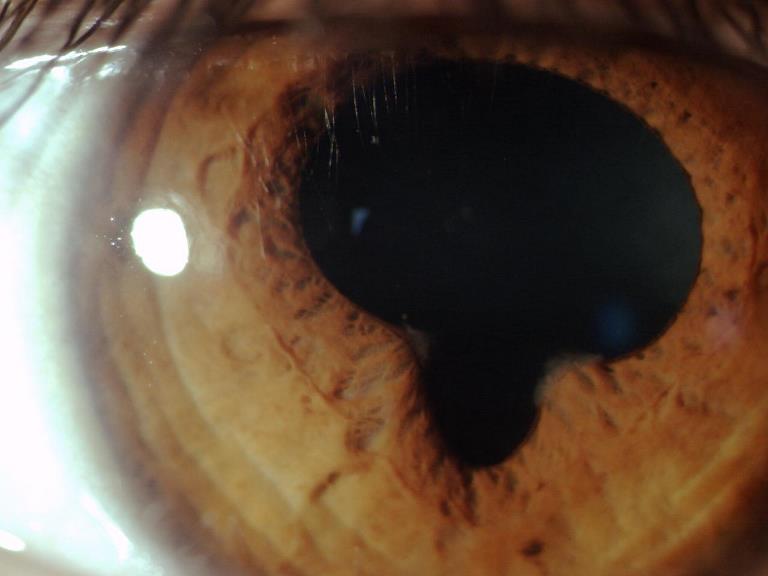
Aderenze che si formano tra l’iride e le altre strutture della camera anteriore dell’occhio in seguito a processi infiammatori che coinvolgono l’iride. Le sinechie anteriori si evidenziano tra l’iride e la superficie posteriore della cornea, provocano spesso la formazione di opacità corneali nella zona di contatto tra le due superfici e occlusione dell’angolo camerulare, con conseguente aumento della pressione oculare.
Le sinechie inoltre possono portare alla deformazione del forame pupillare che può presentarsi stirato in varie direzioni e generare disturbi visivi e abbagliamento.
Se queste ultime si estendono a 360° si può determinare un’occlusione del forame pupillare che impedisce all’umore acqueo di essere drenato correttamente, provocando quindi l’insorgenza di un glaucoma acuto secondario.
Le sinechie posteriori invece si localizzano tra la parte posteriore dell’iride e la faccia anteriore del cristallino, provocando spesso cataratta o glaucoma da occlusione della pupilla.
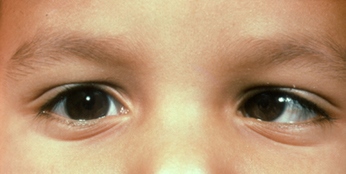
Patologia che consiste nella condizione in cui gli assi visivi dei due occhi non sono allineati. Ciò può comportare diplopia se insorge in età adulta, quando invece è congenito il cervello del bambino esclude spontaneamente l’occhio deviato proprio per evitare la visione doppia: l’occhio non utilizzato in questo caso si definisce “ambliope”.
In base alla direzione della deviazione dell’orientamento lo strabismo può essere convergente (verso l’interno), divergente (verso l’esterno) o verticale (verso l’alto).
Lo strabismo convergente può essere di tipo:
Lo strabismo divergente, molto meno frequente e riscontrabile soprattutto nel bambino in età scolare e nell’adulto. A differenza di quello convergente si manifesta spesso in modo intermittente e varia secondo che il soggetto guardi lontano o vicino. Può essere:
Lo strabismo verticale, invece, è generalmente associato ad una deviazione orizzontale ed è spesso causato da paralisi di origine ostetrica o neonatale.
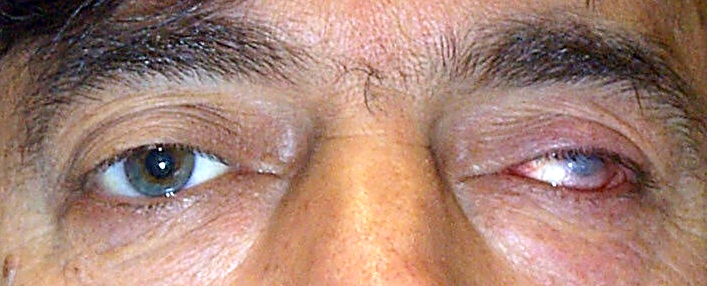
Riduzione patologica del volume del bulbo causata dall’interruzione del flusso ematico o dalla perdita di sostanza interna. Il bulbo può diventare atrofico in seguito a postumi di un trauma, di una ferita perforante o in seguito a patologie come emorragia retinica, panoftalmite od altri processi infettivi. L’occhio subatrofico è quasi sempre non vedente ma spesso non è in quiete e provoca dolore ed infiammazione continua.
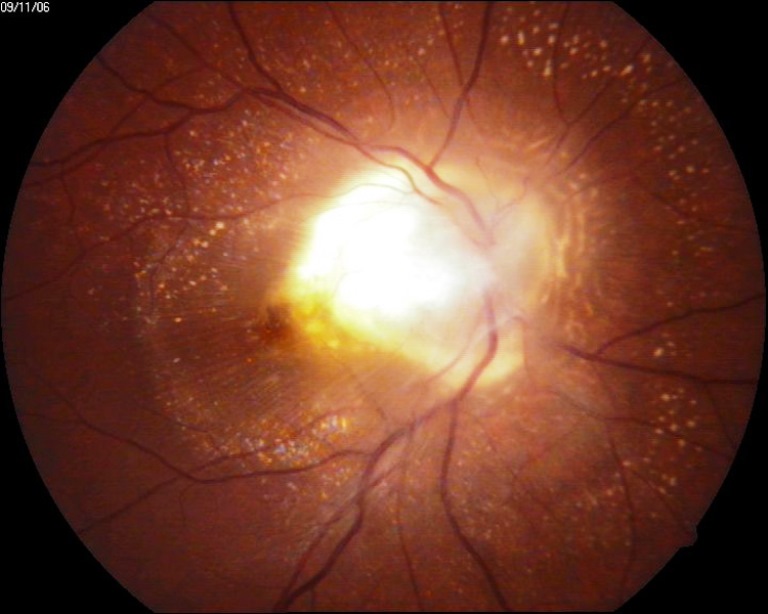
Patologia causata dalla toxocara, un nematode parassita che risiede nel piccolo intestino di cani, gatti e carnivori selvatici e he produce ogni giorno centinaia di uova dove gli embrioni maturano e raggiungono lo stadio larvale infettivo. La prevalenza delle uova di Toxocara nel suolo è correlata al numero di cani presenti in quella zona ed a fattori climatici del luogo.
L’infezione umana è dovuta all’accidentale ingestione di uova infette e la trasmissione può avvenire mediante l’ingestione di cibo contaminato o di terra, per cui i bambini di età inferiore ai 10 anni sono più soggetti ad entrambi i tipi di infezione per la vicinanza con il terreno e per il più frequente contatto con cuccioli di cane.
L’infezione umana può presentarsi in maniera asintomatica, simile ad un’infezione virale e l’incidenza della toxocariasi oculare è anch’essa più frequente nei bambini e costituisce l’1-2% delle uveiti in questo gruppo d’età.
La Toxocara può essere considerata come un possibile agente causale delle uveiti posteriori e di quelle diffuse infantili, l’età media dei pazienti alla diagnosi di toxocariasi oculare è infatti di 7,5 anni e l’80% dei pazienti presentano un’età inferiore ai 16 anni.
Possono essere riscontrate tre differenti manifestazioni oculari: l’endoftalmite cronica, il granuloma posteriore e il granuloma periferico.
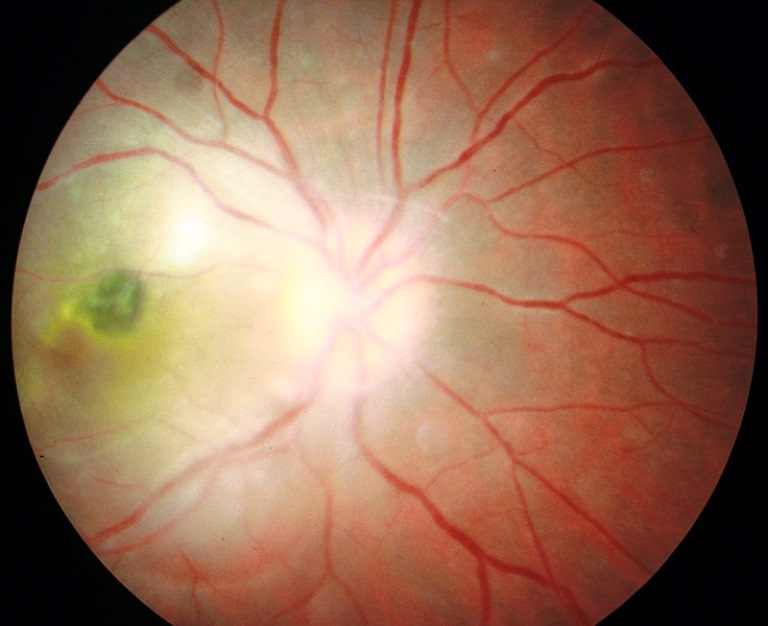
Patologia spesso congenita causata dal protozoo Toxoplasma Gondii presente nelle feci degli uccelli e dei mammiferi come il gatto, nei rettili, nei molluschi ma rilevabile anche nella carne cruda e nelle verdure non lavate accuratamente. Il parassita predilige il tessuto nervoso e può attraversare la placenta durante l’infezione della madre, provocando in determinate circostanze malformazioni o addirittura l’aborto o la morte in utero.
La toxoplasmosi primaria può essere complicata da sintomi gravi come la corioretinite oltre a sintomi attribuibili a una malattia autoimmune. Quest’ultima eventualità è frequente nei malati di Aids o nei soggetti trapiantati, per i quali spesso l’evoluzione è drammatica perché la risposta alla terapia è insufficiente.
La toxoplasmosi acquisita è un’infezione primaria contratta sia con l’ingestione di ovocisti tramite cibo (carni bovine e suinem crude o non sufficientemente cotte, verdure crude non lavate), sia più raramente per ingestione a seguito del contatto con oggetti, terreno o altro materiale contaminato dalle ovocisti di toxoplasma.
L’infezione decorre generalmente in forma asintomatica o con sintomi lievi caratterizzati da interessamento linfoghiandolare. L’interessamento oftalmico della toxoplasmosi provoca l’uveite posteriore infettiva con possibili formazioni di cicatrici a livello retinico ma l’infezione può causare anche gravi patologie come calcificazioni endocraniche, idrocefalo, ritardo mentale ed epilessia.
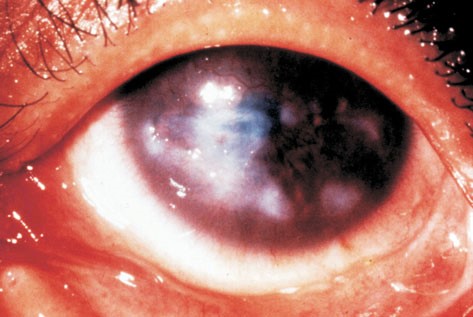
Grave patologia oculare dovuta all’infezione provocata dal batterio Chlamydia Tracomatis. I primi sintomi si manifestano con arrossamento, fotofobia, forte bruciore, lacrimazione ed edema palpebrale. Segno tipico di questa patologia è la palpebra rivolta verso l’interno che si salda alla congiuntiva (entropion cicatriziale) e con essa la trichiasi delle ciglia ed il lagoftalmo, cui conseguono cicatrici corneali fino alla formazione del panno corneale.
L’infezione si trasmette attraverso secrezioni oculari infette oppure indirettamente in seguito a contatto con materiale contaminato ma anche le mosche che si nutrono di secrezioni oculari possono trasmettere la malattia.
Il tracoma è la principale causa di cecità tra le malattie contagiose ed è endemico in alcune zone dell’Africa, del Medio Oriente, dell’India e del Sud-Est asiatico, prediligendo i bambini nei primi due anni di vitae di conseguenza le donne, giustificando l’incidenza due o tre volte più alta della forma più grave cicatriziale nel sesso femminile rispetto a quello maschile.
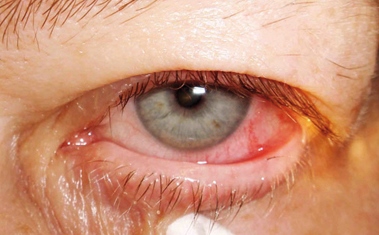
Patologia delle ciglia che deviano dal loro percorso naturale e crescono in direzione del bulbo, venendo a contatto con la cornea e abradendola. Spesso associata all’entropion, si riscontra anche nelle patologie palpebrali croniche ed in seguito a traumi od esiti cicatriziali. La trichiasi che nella sua fase matura è molto dolorosa, deve essere corretta chirurgicamente perché a lungo andare può causare danni corneali irreversibili.
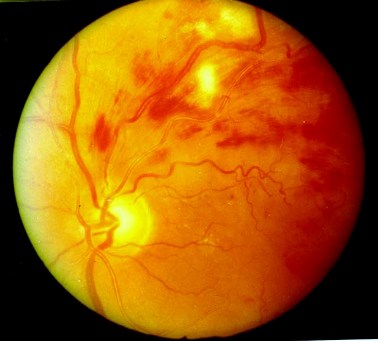
Patologia vascolare che consiste nell’occlusione dell’arteria centrale retinica e che può provocare in pochi minuti lesioni irreversibili alla funzione visiva. L’occlusione può essere dovuta a embolia (placche arterosclerotiche disseminate, endocardite, emboli grassosi) oppure a trombosi di un’arteria centrale sclerotica. Un’altra importante causa è l’arterite cranica. L’occlusione di un ramo dell’arteria retinica è quasi sempre di origine embolica.
E’ imperativo il trattamento immediato e se l’occlusione non viene rapidamente rimossa si verifica un infarto retinico e ne può conseguire la cecità permanente, determinando anche frequentemente la tisi del bulbo che in seguito al mancato afflusso sanguigno entra in una condizione di ipotonia e si atrofizza.
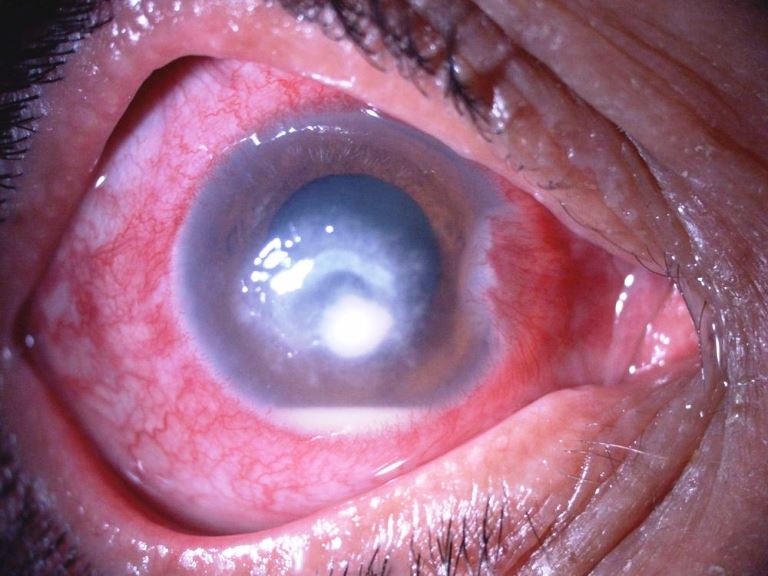
Ulcera batterica
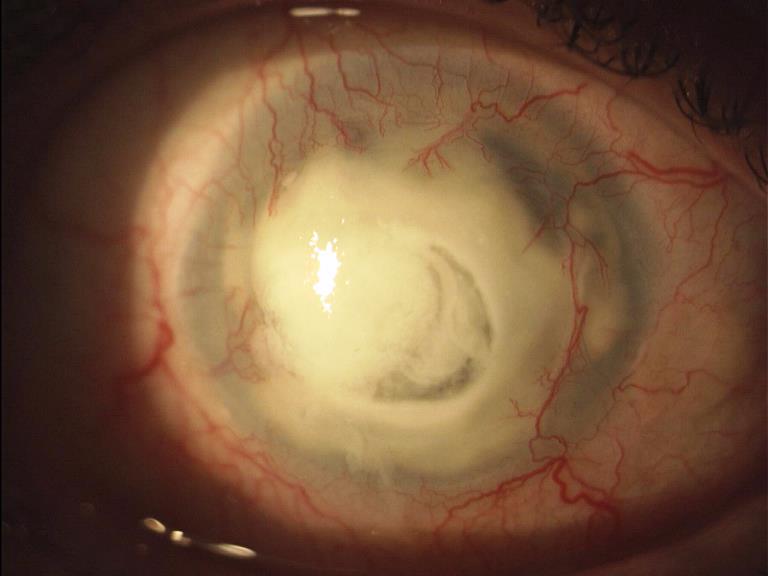
Neovasi cicatriziali
Ulcera infetta
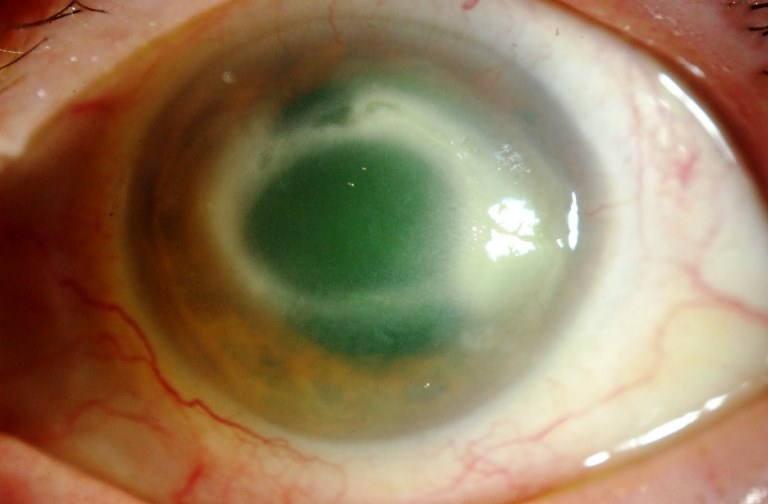
Lesione della cornea in seguito ad un processo infettivo: assume l’aspetto di un piccolo cratere con necrosi locale del tessuto corneale, dovuta all’invasione da parte di batteri, funghi, virus o Acanthamoeba. Se trascurata può mettere in pericolo le altre strutture oculari.
L’ulcera corneale batterica è provocata più frequentemente da Stafilococco, Pseudomonas, o Streptococcus pneumoniae. L’infezione è causata spesso dall’uso delle lenti a contatto durante il sonno, dalla loro inadeguata sterilizzazione ma anche da un trauma corneale oppure da un corpo estraneo corneale.
L’ulcera corneale può anche essere la complicanza di una cheratite neurotrofica, blefarite cronica, congiuntivite (specialmente batterica), tracoma, cheratopatia bollosa e pemfigoide cicatriziale. Può essere anche la conseguenza di alterazioni del trofismo corneale secondarie a carenza di vitamina A o di malnutrizione proteica ed ancora da anomalie palpebrali come l’entropion, la trichiasi, il lagoftalmo e l’esoftalmo).
L’ulcera causate da funghi che sono più croniche rispetto a quelle batteriche, si infiltrano profondamente e possono presentare alla periferia distinte zone di infiltrazione.
Le ulcere corneali provocate da Acanthamoeba, invece, sono molto dolorose e possono manifestarsi con difetti transitori dell’epitelio corneale, multipli infiltrati stromali e successivamente con un unico infiltrato opaco anulare.
L’ulcera erpetica (o dendritica) è provocata dal virus Herpes Simplex o Varicella Zoster in soggetti che hanno subito una prima infezione precedentemente con o senza manifestazioni di herpes labiale o palpebrale. È di norma monolaterale, ha tendenza recidivante e può anche essere concomitante a rash cutaneo di origine erpetica. Questo tipo di ulcera associata a cheratite da herpes simplex è molto pericolosa perché può essere particolarmente refrattaria a qualsiasi trattamento terapeutico.
I sintomi tipici dell’ulcera corneale sono il dolore, senso di corpo estraneo, fotofobia e lacrimazione ma inizialmente possono essere minimi e facilmente sottovalutati. L’ulcera corneale si manifesta inizialmente come opacità superficiale grigiastra e circoscritta e in seguito va incontro a necrosi e suppurazione provocando un’ulcerazione escavata. È presente un difetto dell’epitelio corneale superficiale che si colora di verde con fluoresceina. È frequente un’accentuata iperemia perilimbare e nei casi cronici dal limbus si formano dei neovasi che si dirigono verso l’ulcerazione (neovascolarizzazione corneale).
L’ulcera si può estendere fino a coinvolgere tutta la superficie corneale o può penetrare in profondità nella camera anteriore dove può comparire un ipopion.
Le complicanze sono in diretta relazione con la profondità dell’ulcera: più questa è profonda, più sono gravi. Le ulcere corneali guariscono con formazione di tessuto cicatriziale che causa opacizzazione della cornea e diminuzione del visus. Si possono verificare, con o senza trattamento, irite, iridociclite, perforazione corneale con prolasso dell’iride, ipopion, panoftalmite e perdita dell’occhio.
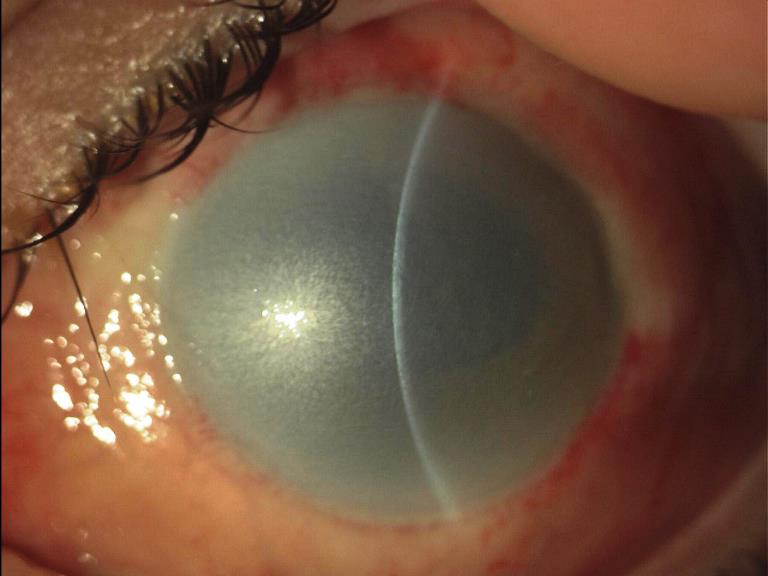
Le causticazioni o ustioni chimiche provocano gravi reazioni tossiche del bulbo. Quelle associate ad un’alterazione del PH possono indurre danni permanenti come risultato del contatto con l’agente chimico.
Le lesioni dovute ad ammoniaca ed altre sostanze alcaline sono le più gravi per la loro rapida diffusione endobulbare che spesso in pochi minuti causano danni oculari permanenti come ulcere profonde con perdita di epitelio corneale e successiva necrosi della congiuntiva.
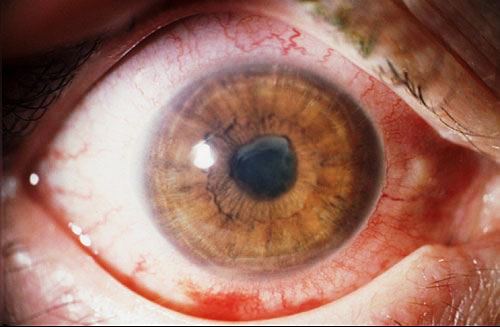
Patologia oculare dovuta ad un processo infiammatorio che interessa l’uvea, membrana costituita da iride, corpo ciliare e coroide. In rapporto alla zona del bulbo dove ha origine l’uveite può essere:
In base al decorso clinico viene classificata in uveite acuta o cronica e in base all’eziologia in uveite esogena (provocate da agenti provenienti dall’esterno) o endogena(da agenti già presenti nel nostro organismo). Può essere inoltre causata da virus, batteri, funghi e parassiti (uveite infettiva) o anche in forma non infettiva in associazione ad altre patologie sistemiche.
I sintomi dipendono dalla sede colpita dall’infiammazione:
Complicazioni:
Le forme anteriori si possono complicare con la formazione di sinechie tra l’iride e la superficie anteriore del cristallino e quando si formano attorno alla pupilla possono provocare il mancato passaggio di umore acqueo dalla camera posteriore del bulbo oculare a quella anteriore, con conseguente aumento della pressione oculare. Altre complicazioni dell’uveite anteriore sono la cataratta e le sofferenze corneali.
L’uveite intermedia ha come complicazioni l’edema maculare cistoide ed il distacco di retina per trazione.
Nell’uveite posteriore le complicazioni riguardano il coinvolgimento della macula da parte dell’infiammazione che provoca edema maculare, fenomeni di occlusione dei vasi della retina, formazione di neovasi coroideali e infiammazione del nervo ottico.
L’uveite è una patologia insidiosa che va trattata con tempestività poiché le forme più gravi possono portare alla cecità.
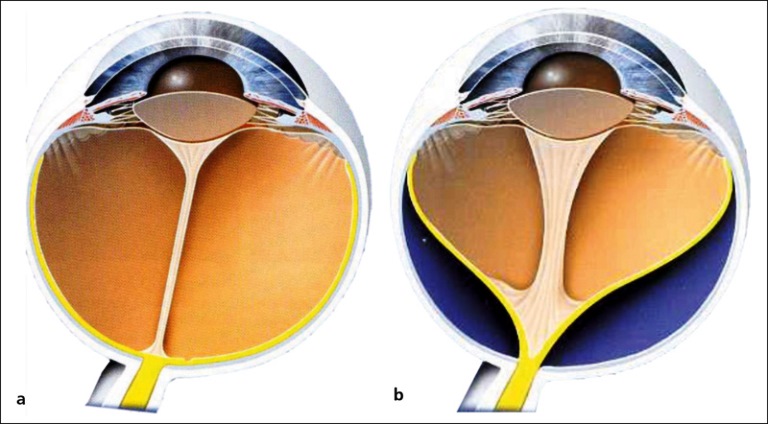
Bulbo normale (a) e con Vitreo primitivo iperplastico (b)
La persistenza del VPI è una rara patologia congenita rara caratterizzata dalla mancata regressione del vitreo primario e della vascolarizzazione vitreale normalmente presente solo nel feto. Può essere distinto in forma anteriore, posteriore e completa a seconda delle strutture oculari coinvolte.
La forma anteriore corrisponde a tessuto fibrovascolare adeso al cristallino e ai processi ciliari. Quella posteriore consiste in un cordone fibrovascolare vitreale che emerge dal nervo ottico e decorre anteriormente. Nella forma completa i due quadri si associano.
Ciò accade perché nella crescita prenatale il segmento posteriore dell’occhio è ricco di rami dell’arteria ialoide proveniente dal nervo ottico (vitreo primario). Successivamente questi vasi sanguigni scompaiono per essere sostituiti dal vitreo secondario che a sua volta origina il vitreo trasparente e avascolare tipico di ogni persona vivente.
Se alcuni vasi sanguigni permangono tipicamente nell’arteria ialoide che porta dal disco ottico alla superficie posteriore del cristallino, permane anche il vitreo primario.
L’occhio con vitreo iperplastico primitivo persistente è frequente nei nati prematuri affetti da Retinopatia del Pretermine (R.O.P.), ed è di solito più piccolo del controlaterale normale (microftalmo o ipoplasia). Molto frequentemente questa patologia si associa a distacco di retina, cataratta e glaucoma, tutti nella forma congenita.
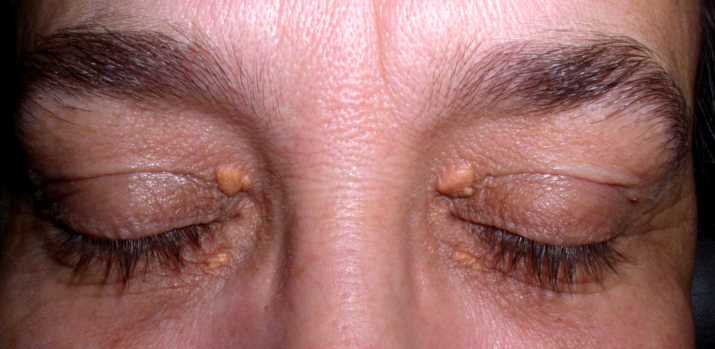
E’ un accumulo di grassi, in particolare di colesterolo localizzato sulle palpebre. Particolarmente diffuso dopo i 40 anni, colpisce di frequente anche i giovani e si presenta come una placca molle e giallastra di forma e dimensione variabile che sporge sulla palpebra. Non causa dolore e non interferisce con la vista. Lo xantelasma può essere rimosso chirurgicamente ma spesso si riforma spontaneamente.

Patologia del bulbo causata da carenza di vitamina A, il cui sintomo caratteristico è la sindrome dell’occhio secco. Può degenerare in ulcera corneale e cheratomalacia.
E’ la maggiore causa di cecità infantile nei Paesi in via di sviluppo. Si stima che 250 milioni di bambini in età prescolare soffrano di carenza di Vitamina A, ogni anno 350.000 bambini diventino ciechi e sempre ogni anno muoiano 2 milioni di bambini per questa patologia.
E’ dovuta alla perdita delle cellule congiuntivali caliciformi e mucosecernenti e ad un’alterazione delle cellule epiteliali congiuntivali. Queste alterazioni provocano la cheratinizzazione dell’epitelio congiuntivale che diviene secco (xerosi congiuntivale). La xerosi si manifesta a livello della congiuntiva bulbare con mancanza di umidità della mucosa, suo ispessimento e raggrinzimento, perdita della pigmentazione e della trasparenza.
La secchezza corneale e congiuntivale può portare a ulcerazione e cheratomalacia con una conseguente perdita della vista nel 50% dei pazienti non trattati. L’ulcerazione corneale sembra essere dovuta al trauma e alla mancanza di proteine associata a una carenza di vitamina A.
Copyright© 2019. Tutti i diritti sono riservati.
Sono vietate le riproduzioni delle immagini e del testo, anche parziali.